


La aproximación terapéutica en fibrosis pulmonar idiopática ha cambiado sustancialmente en los últimos 5 años. Las guías nacionales e internacionales para el tratamiento farmacológico recomiendan el uso de 2fármacos antifibróticos, nintedanib y pirfenidona, con un nivel elevado de evidencia científica a favor del beneficio de ambos fármacos, no solo porque enlentecen la progresión de la enfermedad sino también porque reducen el riesgo de muerte anual. Actualmente, dentro del manejo terapéutico de estos pacientes, se pone en valor tanto el uso de fármacos que actúan sobre mecanismos patogénicos de la enfermedad como el efecto positivo de mejorar la calidad de vida mediante un apoyo integral multidisciplinar que incluye nutrición, actividad física, educación, apoyo emocional y paliación de síntomas. Todo ello buscando que el paciente esté lo mejor posible el mayor tiempo posible desde el diagnóstico. Pero el objetivo de las nuevas terapias antifibróticas combinadas que en estos momentos se evalúan en ensayos clínicos persigue aumentar el beneficio terapéutico o parar completamente la progresión de la enfermedad mediante el potencial efecto sinérgico antifibrótico, al actuar sobre diferentes vías patogénicas a la vez. Finalmente, se investiga qué marcadores podrían ser útiles para identificar pacientes que se puedan beneficiar más de unos fármacos antifibróticos u otros, lo que permitiría optimizar recursos e iniciar los primeros pasos hacia una medicina de precisión en fibrosis pulmonar. A continuación, se revisan los principales puntos de mejora en el tratamiento farmacológico de la fibrosis pulmonar idiopática futuribles a corto, medio y largo plazo.
The therapeutic approach in idiopathic pulmonary fibrosis has changed substantially over the past 5 years. National and international guidelines for the pharmacological treatment of IPF recommend 2antifibrotic drugs, nintedanib and pirfenidone. The use of both these drugs is supported by high-level evidence, with benefits including not only slower disease progression but also a reduction in the annual risk of death. Currently, the therapeutic management of these patients prioritizes both the use of drugs that act on the pathogenic mechanisms of the disease, and the positive effect of improving quality of life with integrated multidisciplinary support, including nutrition, physical activity, education, emotional support, and palliation of symptoms. The overall aim is to ensure that the patient remains as well as possible for as long as possible after diagnosis. However, the goal of the new antifibrotic combinations that are currently under evaluation in clinical trials is to use the potential antifibrotic synergy to enhance the therapeutic benefit or completely halt disease progression, by acting simultaneously on different pathogenic pathways. Another line of investigation involves markers that might be useful for identifying patients who may benefit more from certain antifibrotics than from others, which would make it possible to optimize resources and take the first steps toward precision medicine in pulmonary fibrosis. Below, we review the main potential areas for improvement in the pharmacological treatment of idiopathic pulmonary fibrosis in the short, medium, and long term.
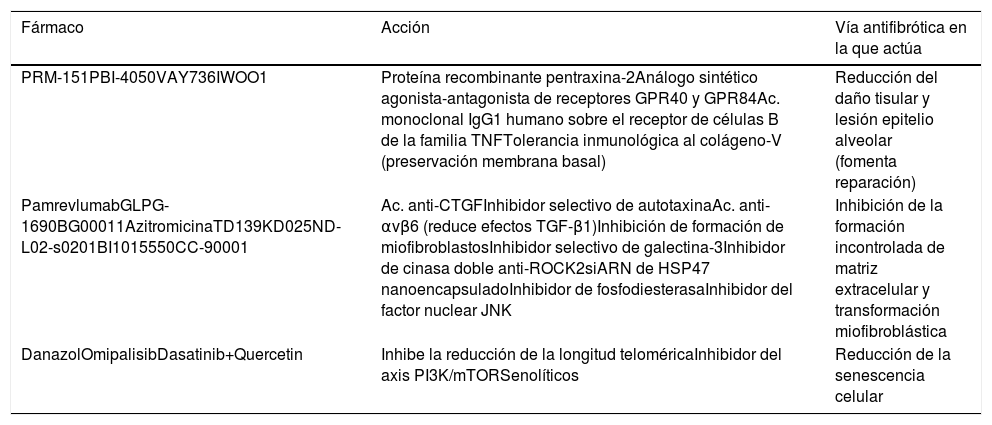
Los pacientes que actualmente se diagnostican de fibrosis pulmonar idiopática (FPI) cuentan con 2opciones farmacológicas que modifican el curso natural de la enfermedad: pirfenidona y nintedanib1-9. Ambas enlentecen la caída de la capacidad vital forzada (CVF) y reducen el riesgo de muerte al año, lo que representa un gran avance1-9. Sin embargo, el siguiente paso sería parar por completo la progresión de la enfermedad y logar que la FPI no sea sinónimo de enfermedad mortal10,11. De forma similar al escenario de hace décadas para enfermedades neoproliferativas hematológicas como leucemia o linfoma, o infecciosas como HIV, con diferentes vías patogénicas implicadas, tal como pasa en la FPI, solo la combinación de fármacos sinérgicos que actúen sobre los diferentes mecanismos permitirá controlar totalmente la enfermedad11,12. Tanto pirfenidona como nintedanib son fármacos pleitrópicos que inhiben diferentes componentes profibróticos3-9. Para ambos fármacos se ha aprendido a optimizar la adherencia y reducir los efectos secundarios, especialmente con un tratamiento multidisciplinar del paciente en la mayoría de los centros expertos, que fomenta su empoderamiento y mejora el control nutricional, emocional y la promoción de la actividad física13. Otros fármacos antifibróticos han demostrado su efecto experimental como inhibidor de fibrosis pulmonar y actualmente su potencial beneficio en pacientes está siendo evaluado mediante ensayos clínicos (tabla 1, fig. 1). Además, no todos los pacientes con FPI se comportan igual clínicamente ni tienen características biológicas idénticas, por lo que el efecto de los fármacos puede ser variable en función de las vías patogénicas predominantes en cada caso11,12. Reconocer a aquellos pacientes con mejor respuesta terapéutica a unos fármacos u otros permitirá optimizar el tratamiento y mejorar su efectividad12.
Nuevos fármacos antifibróticos en ensayo clínico en función de la diana terapéutica y mecanismo profibrótico sobre el que actúa
| Fármaco | Acción | Vía antifibrótica en la que actúa |
|---|---|---|
| PRM-151PBI-4050VAY736IWOO1 | Proteína recombinante pentraxina-2Análogo sintético agonista-antagonista de receptores GPR40 y GPR84Ac. monoclonal IgG1 humano sobre el receptor de células B de la familia TNFTolerancia inmunológica al colágeno-V (preservación membrana basal) | Reducción del daño tisular y lesión epitelio alveolar (fomenta reparación) |
| PamrevlumabGLPG-1690BG00011AzitromicinaTD139KD025ND-L02-s0201BI1015550CC-90001 | Ac. anti-CTGFInhibidor selectivo de autotaxinaAc. anti-αvβ6 (reduce efectos TGF-β1)Inhibición de formación de miofibroblastosInhibidor selectivo de galectina-3Inhibidor de cinasa doble anti-ROCK2siARN de HSP47 nanoencapsuladoInhibidor de fosfodiesterasaInhibidor del factor nuclear JNK | Inhibición de la formación incontrolada de matriz extracelular y transformación miofibroblástica |
| DanazolOmipalisibDasatinib+Quercetin | Inhibe la reducción de la longitud teloméricaInhibidor del axis PI3K/mTORSenolíticos | Reducción de la senescencia celular |

Por último, el tratamiento reparativo o curativo en fibrosis pulmonar es una asignatura pendiente a largo plazo, aunque existen ya resultados experimentales prometedores sobre regeneración tisular en pulmón lesionado14-16 (fig. 2).
Optimización del tratamiento actual en fibrosis pulmonar idiopática
El tratamiento del paciente con FPI no debería limitarse solo a prescribir nintedanib o pirfenidona13. La adherencia y mantenimiento farmacológico depende de varios factores, incluyendo el grado de información y educación que el paciente recibe sobre la enfermedad, los potenciales efectos de los fármacos y cómo prevenir-tratar precozmente los posibles efectos secundarios13. Antes de iniciar el tratamiento se debe valorar el estadio de la enfermedad, los factores pronósticos y las comorbilidades del paciente, así como la situación clínica y familiar1,2. El abanico terapéutico resulta de una aproximación holística o integral que incluye tratamiento sintomático, tratamiento farmacológico antifibrótico, evitar factores que puedan agravar la enfermedad (sobre todo infecciones víricas y reflujo gastroesofágico), manejo de comorbilidades (enfisema-obstrucción de vía aérea, hipertensión pulmonar), ejercicio de fisioterapia-rehabilitación para mantener calidad de vida y autonomía, cuidado emocional y psicológico, oxigenoterapia cuando se requiera y tener siempre presente el trasplante pulmonar en los casos que cumplan los criterios13. Por último, en la fase avanzada o final de la enfermedad, tratamiento paliativo en coordinación con los servicios de atención a domicilio1,2. Idealmente, en el tratamiento de la FPI interviene un equipo multidisciplinar que engloba neumólogos, farmacéuticos, enfermería especializada, fisioterapia-rehabilitación y apoyo psicológico, con el objetivo de cubrir las necesidades prioritarias1,2.
Pirfenidona es un fármaco que reduce el factor transformador de crecimiento β (TGF-beta1) y el factor de crecimiento plaquetario (PDGF)1,3,4. Su eficacia ha sido evaluada en pacientes con FPI leve-moderada (CVF ≥ 50%, DLCO ≥ 30-35%) mediante ensayos clínicos aleatorizados, doble ciego y controlados con placebo (CAPACITY-1, CAPACITY-2, ASCEND, SP2, SP3)1,3,4, y también su seguridad a largo plazo (ensayo clínico PASSPORT). Los efectos beneficiosos de este fármaco, entre los que destacan un menor deterioro de la CVF y una reducción significativa del riesgo relativo de muerte al año del 48%4, son independientes de la CVF al inicio5. La dosis recomendada es de 2.403mg/día, dividida en 3tomas, durante las comidas, que se alcanza de forma incremental en unas 3 semanas tras comprobar analíticamente la tolerancia hepática1,3,4. Los efectos adversos más relevantes son gastrointestinales (náuseas, dispepsia y pérdida de peso) seguido de rash, fotosensibilidad y, en menor proporción, alteraciones hepáticas3,4. La mayoría de estos efectos pueden ser prevenibles, en caso de presentarse suelen ser manejables y aparecen más a menudo en los primeros 6 meses3,4. Actualmente se está ensayando (fase I) una nueva formulación nebulizada de pirfenidona (100mg) que pretende reducir los efectos secundarios al conseguir una mayor concentración sistémica del fármaco (fig. 1).
Nintedanib es un inhibidor triple de los receptores tirosina cinasa que presenta actividad anti-PDGF, antifactor de crecimiento de los fibroblastos (FGF y antifactor de crecimiento vascular endotelial)6-8. Ensayos aleatorizados, doble ciego y controlados con placebo avalan la utilidad clínica del fármaco en FPI leve-moderada (TOMORROW, INPULSIS-1 e INPULSIS-2)6,7. Nintedanib, a dosis de 150mg/12h, inhibe el deterioro de la CVF, alarga el tiempo hasta la primera agudización grave y reduce el riesgo de muerte a un año6-8. Su efecto beneficioso es independiente de la existencia o no de enfisema y del valor de CVF al inicio9. La aparición de diarrea es frecuente (50-60%), pero suele ser leve o moderada y controlable con dieta astringente, probióticos, ultralevura y en, ocasiones, loperamida o codeína6-8. Otros efectos adversos menos frecuentes son pérdida de peso y alteraciones en la función hepática6,7. Nintedanib está contraindicado en pacientes con reacciones alérgicas al cacahuete o a la soja6,7. El uso de los anticoagulantes orales más comunes, como sintrom o warfarina, no ha sido testado en los ensayos clínicos y podría incrementar el riesgo de hemorragia17. En casos que requieran anticoagulación, se aconseja valorar el uso de heparinas o anticoagulantes orales de nueva generación17. La seguridad a largo plazo ha sido testada en ensayo clínico (INPULSIS-ON).
No existen estudios que evalúen el efecto de estos fármacos en función de las vías patogénicas profibróticas predominantes y, por lo tanto, del fenotipo del paciente. Sin embargo, actualmente se está llevando a cabo un ensayo clínico (INMARK) para evaluar el efecto biológico de nintedanib en pacientes con FPI leve (CVF > 80%) e identificar el tipo de paciente que más se beneficia de este tratamiento18. Por otro lado, un reciente estudio post hoc de los ensayos clínicos CAPACITY-1, 2 e INPULSIS demuestra que pirfenidona tiene el mismo beneficio clínico en pacientes con y sin acortamiento telomérico19, aunque la progresión rápida de la enfermedad sea más frecuente en pacientes con acortamiento severo.
Tratamiento antifibrótico en combinación: parar progresiónEl uso de diferentes líneas de tratamiento simultáneamente podría actuar de forma sinérgica y generar un mayor beneficio clínico que la monoterapia, dada la complejidad de los mecanismos patogénicos que intervienen10-12. El avance del conocimiento sobre la patogenia de la enfermedad ha hecho posible identificar dianas terapéuticas sobre las que actuar para frenar la progresión (factores de crecimiento y células implicadas) e incluso pararla al intentar modificar la respuesta reparativa alterada (fenómenos comunes al envejecimiento acelerado)14. La opción de combinar más de un fármaco antifibrótico es posible a través de ensayos clínicos que actualmente están en marcha (fig. 1).
Hasta la fecha se han finalizado escasos ensayos clínicos que combinan 2fármacos. Al añadir el antioxidante N-acetilcisteína a la pirfenidona, se ha observado un incremento de los efectos adversos digestivos, sin aumentar el beneficio terapéutico20. El uso combinado de nintedanib con sildenafilo en pacientes con FPI severa (DLCO < 35%) tampoco ha objetivado mayor beneficio21. Este ensayo clínico (INSTAGE) tuvo como objetivo primario evaluar el efecto sobre la calidad de vida21 y, aunque resultó negativo, se está analizando el efecto de ambos fármacos sobre otras variables, especialmente en el subgrupo de pacientes con mayor afectación cardíaca derecha. El estudio permitió reconocer que el tratamiento con nintedanib en FPI grave conseguía valores de caída de CVF (mL) similares a los casos leves-moderados incluidos en INPULSIS21. Recientemente se ha finalizado el ensayo clínico que combina pirfenidona y nintedanib en FPI grave con hipertensión pulmonar, pero está pendiente la presentación de resultados22. Por otro lado, 2ensayos clínicos fase II que combinan pirfenidona y nintedanib han demostrado que el tratamiento es seguro y no aumenta el porcentaje de efectos adversos graves23,24. Además, al añadir nintedanib a la pirfenidona (estudio INJOURNEY) hubo un incremento no significativo de la CVF a las 8 semanas de tratamiento23.
Por otro lado, existen otros ensayos clínicos con nuevos antifibróticos que evalúan; 1) la seguridad farmacológica (ensayos fase I y fase II) y 2) el beneficio en FPI con su administración en monoterapia o en combinación de estos con pirfenidona o nintedanib (ensayos fase II y fase III) (tabla 1, fig. 1; https://clinicaltrials.gov). Pamrevlumab es un anticuerpo monoclonal contra el factor de crecimiento de tejido conectivo (anti-CTGF), que fue bien tolerado en ensayo clínico fase II (PRAISE). Testado a dosis de 30mg/kg y administrado cada 3 semanas, los resultados demostraron que en monoterapia o asociado a nintedanib o pirfenidona es un fármaco seguro que reduce no significativamente la caída de CVF y el deterioro de la calidad de vida (USCD-SOBQ)25.
GLPG1690 es un nuevo compuesto oral, inhibidor selectivo de la autotaxina, que reduce la migración celular y la producción de tenascina-C y colágeno26. El axis autotaxina-fosfatasa lisofosfatídica fosfato se encuentra incrementado en FPI y GLPG1690 inhibe pleiotrópicamente la fibrosis pulmonar in vivo e in vitro. Los resultados del ensayo clínico fase II FLORA mostraron que GLPG1690 (600mg/día) es seguro y además permite hacer un seguimiento del efecto biológico al reducir las concentraciones séricas de axis autotaxina-fosfatasa lisofosfatídica fosfato26. Actualmente GLPG1690 está siendo evaluado en combinación con nintedanib o pirfenidona, en el ensayo clínico fase III ISABELA.
BG-00011 (STX-100) es un anticuerpo monoclonal específico anti-αvβ6, que bloquea la fosforilación de SMAD2 en lavado broncoalveolar27. El ensayo clínico fase iib demostró seguridad del fármaco y actualmente está en la fase III.
PRM-151 es una forma recombinante de la proteína endógena humana pentraxina 2 (PTX-2), la cual se activa para reparar el daño tisular28. Este agonista regula la diferenciación macrofágica. En fase I y II, PRM-151 (10mg/kg al día por vía intravenosa) ha demostrado buena tolerancia y seguridad, y los resultados exploratorios sobre la CVF y DLCO son alentadores28.
Azitromicina es un macrólido con actividad inmunomoduladora que puede disminuir el daño pulmonar e inhibir la transformación miofibroblástica29. Actualmente está en marcha un ensayo fase II para evaluar su efecto (500mg/día vía oral) sobre la tos y la función pulmonar (CVF) (NCT02173145).
PBI-4050 es un análogo sintético con afinidad agonista y antagonista a receptores-G-proteicos, que regula la actividad de macrófagos, fibroblastos/miofibroblastos y células epiteliales30. Un reciente ensayo clínico ha demostrado su seguridad, administrado una vez al día, en 4 cápsulas de 200mg, durante 12 semanas30.
Galectina-3 es una proteína circulante que activa macrófagos y miofibroblastos. TD139 es un inhibidor de galectina-3 que se administra en forma inhalada, en polvo seco31. En un ensayo clínico fase I/II (NCT02257177) TD139ha demostrado ser un fármaco seguro y bien tolerado.
Danazol es un tratamiento hormonal utilizado en la insuficiencia de médula ósea desde el siglo pasado32. Un estudio exploratorio en el que se incluyó a 27 pacientes con acortamiento telomérico y diferentes expresiones clínicas demostró que este fármaco, administrado vía oral (800mg/día), mantenía la longitud telomérica tras 24 meses32. En la actualidad se evalúa su efecto en ensayo clínico fase II en FPI con acortamiento telomérico (NCT03312400).
KD025 (SLX-2119) es un inhibidor ROCK2 que actúa sobre la infiltración macrofágica, activación endotelial y diferenciación miofibroblástica33. Se está desarrollando un ensayo clínico fase II (NCT02688647) para evaluar la seguridad y actividad del fármaco, administrado oralmente (400mg/día) durante 24 semanas.
VAY736 es un anticuerpo que actúa sobre el receptor de células B de la familia TNF (BAFF-R), disminuye las células B activas y bloquea la señal BAFF34. VAY736 se administra de forma subcutánea, cada 4 semanas (NCT03287414).
ND-L02-s0201 (BMS-986263) es una nanopartícula lipídica que encapsula un silenciador siARN de la proteína HSP47 (chaperón específico de colágeno asociado a mal pronóstico)35, que se administra de forma intravenosa (NCT03538301).
BI1015550 es un nuevo inhibidor de la fosfodiesterasa, evaluado en ensayo fase I a dosis incremental vía oral (NCT03422068).
CC-90001 es un inhibidor JNK, con buena tolerancia a dosis de 200mg/día y 300mg/día en un número limitado de pacientes (fase ib) a 12 semanas, con el que se objetiva un incremento en la CVF en el 83% de los casos, así como reducción sérica de tenascina-C (marcador de actividad fibrótica) (NCT03142191).
IWOO1 es un fármaco oral bien tolerado que evita la degradación del colágeno V en pacientes con anticuerpo anticolágeno V positivo y parece estabilizar la CVF (ensayo fase I NCT01199887).
Tratamientos experimentales con objetivo reparativo o curativoUna vez identificados y evaluados los factores fisiológicos que intervienen en la reparación tisular y que, en su mayoría, están en relación con fenómenos de envejecimiento, el siguiente paso es investigar tratamientos que permitan regular estas alteraciones para reparar el tejido lesionado10,11,14,36.
El axis fosfoinositol-3-cinasa (PI3K)/mTOR es fundamental en el metabolismo y ciclo celular, proliferación y autofagia, y su inhibidor, omipalisib (GSK2126458), resulta un potente antifibrótico (fase I)37. El incremento de la senescencia celular se asocia al envejecimiento acelerado identificado en FPI, fundamental en el desarrollo y progresión11,14. El fenotipo secretor senescente observado inicialmente en células epiteliales alveolares y posteriormente en otras estirpes celulares induce la producción de colágeno y otros fenómenos profibróticos38. Por lo tanto, la reducción de la senescencia celular, tanto en forma de senolisis como en forma de rescate de senescencia, resulta un nuevo objetivo terapéutico13,38. La mayoría de los estudios con fármacos senolíticos han sido en modelos animales38. Recientemente un estudio piloto no aleatorizado ha evaluado la seguridad del tratamiento senolítico intermitente semanal (dasatinib 100mg/día + quercetina 1.250mg/día) durante 3 semanas en 14 pacientes con FPI39. Los resultados clínicos y biológicos exploratorios positivos han llevado a la realización de actuales ensayos clínicos (fig. 1).
El acortamiento telomérico acelerado es una característica biológica que se puede identificar en una cuarta parte de los pacientes con FPI13,40. Las mutaciones en genes de la enzima telomerasa se identifican en un porcentaje limitado de los casos (11% en casos esporádicos, 25% en familiares)13,40. Los pacientes que presentan telómeros cortos asocian mayor mortalidad a 3 años, especialmente cuando la enfermedad se desarrolla en edades más jóvenes (por debajo de 60 años), y, además, el mal pronóstico de estos pacientes es independiente del tipo de patrón radiológico fibrosante13,40. Estos pacientes presentan un mayor efecto deletéreo del tratamiento inmunosupresor41, lo que resulta de especial interés en fibrosis pulmonar no idiopática, donde los tratamientos inmunosupresores son habituales. Finalmente, las mutaciones en genes de telomerasa pueden asociar otras enfermedades hematológicas, neoproliferativas, inmunológicas o degenerativas, por lo que este target terapéutico no solo resulta de interés en FPI sino también en otras formas de telomeropatías42. Estudios recientes demuestran que la regulación de la enzima telomerasa reduce la disfunción telomérica y el daño al ADN43,44. GSE4 (gestelmir) es un péptido de la disquerina que inhibe la oxidación y el déficit telomérico, reduce fenómenos de senescencia celular e inhibe la fibrosis pulmonar inducida en modelo animal43. AAV9-Tert es un tratamiento genético que activa la telomerasa, recupera la capacidad proliferativa de células epiteliales alveolares y disminuye la fibrogénesis pulmonar inducida por bleomicina44.
La hormona tiroidea resulta crítica para la homeóstasis celular en respuesta a estrés45. El tratamiento aerosolizado con hormona tiroidea incrementa la supervivencia y mejora la fibrosis inducida en modelo animal al promover la biogénesis y bioenergética mitocondrial y reducir la apoptosis alveolar asociada45.
El proteosoma es un complejo multicatalítico enzimático (20S proteosoma) que lisa los polipéptidos en diferentes aminoácidos, lo que es clave para la limpieza de productos de lisis en el proceso reparativo46. Oprozomib es el primer inhibidor proteosómico oral probado en estudios experimentales que reduce la respuesta fibrogénica pulmonar46.
La vía de señalización Wnt-β-catenina, alterada en fibrosis pulmonar, es relevante en la regulación, diferenciación, proliferación y muerte celular, por lo que afecta al desarrollo reparativo47. Su regulación directa o indirecta a través del efecto sobre exosomas que inducen esta cascada se ha evaluado experimentalmente48.
Diversos estudios han testado células madre mesenquimales y otras células pluripotenciales como posible terapia antifibrótica dados sus potenciales efectos inmunomoduladores, antiinflamatorios y antifibróticos49. La función de estas células depende de diferentes factores, entre ellos principalmente la edad y la existencia de una base biológica reparativa alterada, como pasa en FPI49. Por ello, la mayoría de los estudios han utilizado el trasplante alogénico celular16. Los diferentes ensayos clínicos han evaluado, por vía intravenosa o endotraqueal, diferentes grados de pureza celular y orígenes de las células pluripotencialas variadas (médula ósea, grasa, cordón umbilical, pulmón), diferentes tiempos y pretratamientos, y, de momento, aunque parece una terapia segura, ninguno de los estudios ha podido ser replicable de forma multicéntrica y confrontado a un grupo control paralelo16,50. Entre las limitaciones observadas destacan la dificultad en el aislamiento y pureza celular, la semivida de las células pluripotenciales una vez infundidas y la falta de control en su ciclo16,49. Poco a poco se avanza para superar todas las dificultades51,52: existen opciones que utilizan menos tejido pulmonar para obtener células pluripotenciales esferoidales51, se busca el origen celular con mayor poder reparativo y se simplifica la terapia buscando el beneficio de estas células a través del efecto antifibrótico y reparativo de sus productos (factores de crecimiento como HGF y microvesículas)52.
ConclusiónMás de 200 fármacos antifibróticos han sido desarrollados experimentalmente; la mayoría no llega o no supera al ensayo clínico. Sin embargo, en la actualidad, además de los 2fármacos antifibróticos en monoterapia, 4medicamentos más en ensayo clínico fase III se evalúan en combinación con pirfenidona o nintedanib, intentando controlar mejor o parar la progresión. Posiblemente, a largo plazo seremos capaces de individualizar las terapias combinadas y reparar el daño pulmonar.
FinanciaciónInstituto de Salud Carlos III (ISCIII) PI18/00367, cofinanciado con los fondos FEDER/Fondos Europeos de Desarrollo Regional (ERDF: a way to build Europe).
Conflicto de interesesEl grupo de investigación que lidera la autora recibe o ha recibido fondos en forma de becas o pago por prestación de servicio científico de Roche, Boeringher Ing., Chiesi, Esteve Teijin Healthcare, Linde, GSK, Astra-Zeneca, Intermune.







