







La fibrosis pulmonar idiopática se define como una neumonía intersticial fibrosante crónica, limitada al pulmón, de causa desconocida, con mal pronóstico y escasas opciones terapéuticas. En los últimos años se ha observado un incremento en su prevalencia, probablemente debido a la optimización de los métodos diagnósticos y al aumento de la esperanza de vida. En el consenso ATS/ERS del año 2000 se establecieron por primera vez los criterios diagnósticos y las recomendaciones para evaluar su evolución y tratamiento. Posteriormente, diversos estudios han contribuido a optimizar las pautas diagnósticas y terapéuticas. En el año 2011 se publicó un consenso internacional en el que se redefinieron los criterios diagnósticos y se establecieron nuevas recomendaciones terapéuticas. En esta normativa se actualizan los aspectos novedosos del diagnóstico y el tratamiento de la fibrosis pulmonar idiopática. Se ha atribuido un nivel de evidencia a las cuestiones más relevantes, principalmente en el apartado dedicado al tratamiento.
Idiopathic pulmonary fibrosis is defined as a chronic fibrosing interstitial pneumonia limited to the lung, of unknown cause, with poor prognosis and few treatment options. In recent years there has been an increase in their prevalence, probably due to the optimization of diagnostic methods and increased life expectancy. The ATS/ERS Consensus (2000) established the diagnostic criteria and recommendations for the assessment of the disease course and treatment. Later studies have helped to redefine diagnostic criteria and treatment options. In 2011, an international consensus was published, establishing diagnostic criteria and new treatment strategies. These guidelines have been updated with the newest aspects of diagnosis and treatment of idiopathic pulmonary fibrosis. A level of evidence has been identified for the most relevant questions, particularly with regard to treatment options.
El consenso ATS/ERS publicado en el año 2000 sobre la fibrosis pulmonar idiopática (FPI) estableció por primera vez los criterios diagnósticos y las recomendaciones para evaluar su evolución y tratamiento1. Desde su publicación, diversos estudios han contribuido a optimizar las pautas diagnósticas y terapéuticas de la FPI. Debido a ello, en el año 2011 se publicó un consenso internacional en el que se redefinieron los criterios diagnósticos y se establecieron nuevas recomendaciones terapéuticas2. En 2003, el Grupo de Investigación en Enfermedades Pulmonares Difusas de la SEPAR elaboró la Normativa sobre el Diagnóstico y Tratamiento de las Enfermedades Pulmonares Intersticiales Difusas (EPID)3. Con el propósito de actualizar esta normativa, nos pareció oportuno limitarla a la FPI, ya que es una de las enfermedades en la que se han producido más cambios y avances en los últimos años. En la presente normativa se detallan los cambios que han acaecido en los criterios diagnósticos y en las nuevas estrategias terapéuticas. Para la determinación de las recomendaciones y evidencias se ha utilizado el sistema GRADE4.
DefiniciónLa FPI se define como una neumonía intersticial fibrosante crónica, limitada al pulmón, de causa desconocida, que afecta generalmente a adultos mayores de 50años, y asociada al patrón radiológico y/o histológico de la neumonía intersticial usual (NIU)2.
Incidencia y prevalenciaSe han realizado varios estudios para evaluar la incidencia y la prevalencia de la FPI. Los datos más fiables estiman que la incidencia varía entre 4,6 y 7,4/100.000 habitantes, y la prevalencia se sitúa entre 13/100.000 habitantes en mujeres y 20/100.000 habitantes en varones2. De acuerdo con estos datos, se estima que en España la FPI puede estar afectando a unas 7.500 personas. Se desconoce si la incidencia y la prevalencia están influidas por factores étnicos, raciales o geográficos. En los últimos años se ha observado un incremento en la prevalencia, probablemente debido a la optimización de los métodos diagnósticos y al aumento de la esperanza de vida5,6.
Historia naturalLa historia natural de la FPI es variable e impredecible en el momento del diagnóstico (fig. 1). Algunos pacientes pueden permanecer asintomáticos durante 2-3años. No obstante, la mayoría presentan una lenta progresión con deterioro clínico y funcional que finalmente ocasiona insuficiencia respiratoria crónica. En otros casos existen periodos de relativa estabilidad con episodios de agudización (exacerbaciones agudas u otras complicaciones) que son causa de una alta morbimortalidad2. En una minoría de pacientes la enfermedad es de corta duración, con una progresión más rápida (forma acelerada)7. En general la supervivencia media es de 2-5años desde el inicio de los síntomas2. Se desconoce si las diferentes formas de historia natural representan fenotipos diferentes de la enfermedad.
Etiología y factores de riesgo
La etiología de la FPI no se conoce, aunque probablemente es debida al efecto de diversos factores en sujetos con predisposición genética8.
Factores genéticosLas alteraciones genéticas con más relevancia clínica son: mutaciones en los genes que mantienen la longitud de los telómeros (TERT, TERC), que son más frecuentes en las formas familiares, en la proteína C del surfactante y en la región promotora de la mucina 5B (MUC5B). No existe ninguna prueba genética establecida para valorar la predisposición a la FPI8,9.
Factores ambientalesEl tabaquismo (>20 paquetes/año) y la exposición al sílice, al latón, al acero, al plomo y al polvo de madera, las actividades laborales en ganadería y en agricultura, y la construcción de casas de madera son considerados factores de riesgo8,10,11.
Reflujo gastroesofágicoVarios estudios han demostrado que el reflujo gastroesofágico (RGE) es un factor de riesgo para la predisposición y la progresión de la FPI12.
Infecciones víricasNo existe evidencia suficiente para considerar que las infecciones víricas (virus de la hepatitisC, herpes virus, adenovirus) sean factores etiológicos de la FPI, aunque su contribución sigue siendo objeto de estudio.
AutoinmunidadEl posible origen autoinmune de la FPI se basa en que las manifestaciones radiológicas y/o histológicas de la NIU se asocian a enfermedades del tejido conectivo, aunque estas suelen cursar con histología de neumonía intersticial no específica
DiagnósticoEl diagnóstico definitivo de FPI requiere: a)la exclusión de otras entidades clínicas definidas o enfermedades parenquimatosas pulmonares difusas de causa conocida (exposición ambiental u ocupacional, enfermedades del tejido conectivo, toxicidad por fármacos), y b)la presencia de un patrón histológico de NIU en el examen del tejido pulmonar obtenido mediante biopsia pulmonar quirúrgica, o bien la evidencia radiológica de patrón NIU en la tomografía axial computarizada de alta resolución (TCAR), o ambas.
Una valoración multidisciplinar en el que participen neumólogos, radiólogos y patólogos expertos en el diagnóstico y manejo de las EPID consigue aumentar la precisión diagnóstica, y en el momento actual es una recomendación ampliamente aceptada para establecer el diagnóstico (fig. 2)2,13.

El cuadro clínico de la FPI es de comienzo insidioso y se suele caracterizar por disnea de esfuerzo progresiva, en muchas ocasiones acompañada de tos improductiva. El inicio de los síntomas es lento, pero van empeorando con el tiempo. La demora entre el inicio de la sintomatología y el diagnóstico final es variable y puede estar entre los 6meses y los 2años14. La presencia de síntomas/signos sistémicos debe hacer sospechar un diagnóstico alternativo. Se auscultan estertores crepitantes en el 90% de los pacientes y se objetivan acropaquías en el 50%3.
No hay alteraciones de laboratorio específicas para esta enfermedad. Sin embargo, aun en ausencia de signos o síntomas específicos de enfermedades del tejido conectivo se deben realizar determinaciones serológicas de autoinmunidad en todos los pacientes2. En efecto, se puede detectar positividad en los anticuerpos antinucleares o factor reumatoide hasta en el 20% de los casos de FPI3. Deben valorarse de forma sistemática la presencia de IgG específicas en suero frente a los antígenos que con más frecuencia pueden causar neumonitis por hipersensibilidad, dado que sus manifestaciones clínicas en ocasiones son similares a las de la FPI. En caso de positividad a alguna de ellas, en el contexto de una exposición plausible y lavado broncoalveolar (LBA) con aumento de porcentaje de linfocitos, deberá practicarse una prueba de provocación frente al antígeno en cuestión y/o biopsia pulmonar quirúrgica, con el fin de confirmar o no el diagnóstico de neumonía por hipersensibilidad crónica3. Por otra parte, en los últimos años ha cobrado interés la posibilidad de emplear nuevos biomarcadores en el diagnóstico y la caracterización de esta enfermedad. Algunos —como KL-6, SP-A y SP-D, fibrocitos circulantes y metaloproteinasas1 y 7— están bajo investigación en el momento actual15–18.
Lavado broncoalveolar y biopsia transbronquialEl LBA ha sido ampliamente empleado en el estudio de las EPID. Su análisis en la FPI suele mostrar neutrofilia discreta con o sin eosinofilia, y su empleo clásicamente ha estado relacionado con su capacidad para descartar otras entidades2,3. En el último consenso internacional se señala que el LBA con análisis celular no debería realizarse de forma sistemática a todos los pacientes en el proceso diagnóstico pero, sin embargo, podría ser apropiado para una minoría2. No obstante, en casos determinados el LBA puede ser de gran ayuda en el diagnóstico diferencial con otras entidades, como la neumonitis por hipersensibilidad crónica o la neumonía intersticial no específica.
La biopsia transbronquial es útil en las enfermedades con distribución linfática y centrolobulillar o en las que presentan rasgos diagnósticos característicos y que tienen una distribución difusa19. Con mayor frecuencia se utiliza para el diagnóstico de sarcoidosis, infecciones y tumores19. En cambio, no tiene utilidad en el diagnóstico de la FPI, dado que no es posible observar la distribución de la lesión por el tamaño de la muestra. La incorporación a la práctica de la criobiopsia es muy prometedora, pero se precisan más estudios para corroborar su utilidad en las EPID20.
Tomografía axial computarizada de alta resoluciónLa TCAR representa posiblemente el mayor avance diagnóstico de las 2 últimas décadas en el estudio de las enfermedades pulmonares difusas. La TCAR, ya sea mediante adquisición secuencial («adquisición corte a corte») o volumétrica («adquisición continua»), es la técnica indiscutible en el diagnóstico de la FPI. El conocimiento de la dosis de radiación empleada en la TCAR es muy importante, pues la dosis de radiación empleada en la TCAR volumétrica triplica los valores alcanzados mediante la TCAR secuencial. La decisión entre utilizar una u otra técnica dependerá del balance entre la información esperada y el riesgo individual por el aumento de la radiación recibida. Se recomienda el seguimiento de los protocolos establecidos, teniendo en cuenta que la edad y el sexo del paciente son factores decisivos (p.ej., TCAR secuencial, con intervalos de 10mm, en la valoración inicial de pacientes menores de 40años, y tomografía computarizada multidetector (TCMD) en pacientes de 50años o mayores)21.
El objetivo es identificar los hallazgos típicos del patrón de NIU (fig. 3) y distinguirlos de los patrones menos específicos presentes en las otras neumonías intersticiales idiopáticas. Para evitar errores descriptivos y de concepto, la lectura radiológica debe utilizar una terminología descriptiva basada en la correlación radiológico-patológica, como la recomendada por la sociedad Fleischner22.
.")
El consenso oficial ATS/ERS/JRS/ALAT 20112 establece que, en la TCAR, el diagnóstico de certeza de la NIU se basa en la identificación de los 4 hallazgos «típicos»: a)la afectación pulmonar debe tener un predominio basal y una localización subpleural; b)presencia de reticulación evidente; c)existencia de panalización con/sin bronquiectasias/bronquiolectasias de tracción, y d)demostrar la ausencia de hallazgos considerados excluyentes de un patrón de NIU (tabla 1, fig. 3). La presencia de vidrio deslustrado debe ser mínima o inexistente.
Tomografía axial computarizada de alta resolución (TCAR): hallazgos considerados excluyentes de un patrón de NIU
| Predominio en campos medios o superiores |
| Predominio peri-broncovascular |
| Presencia importante de vidrio deslustrado |
| Numerosos micronódulos (bilaterales, lóbulos superiores) |
| Quistes (múltiples, bilaterales, distantes de las zonas de panalización) |
| Patrón de atenuación en mosaico/atrapamiento aéreo (bilateral, en 3 o más lóbulos) |
| Consolidación segmentaria |
De Raghu et al.2.
La panalización, formada por grupos de quistes con paredes finas, con una localización subpleural y un diámetro de entre 3 y 10mm, es un hallazgo imprescindible para diagnosticar con certeza el patrón de NIU. Cuando no hay panalización visible, el diagnóstico mediante la TCAR será el de posible patrón de NIU; en estos casos, el diagnóstico definitivo de NIU deberá realizarse mediante biopsia. La biopsia pulmonar puede evitarse únicamente cuando la TCAR muestre un patrón de certeza típico de NIU. El valor predictivo positivo de la TCAR en el diagnóstico de la NIU es del 90 al 100%. Un patrón de NIU puede identificarse también en la neumonitis por hipersensibilidad crónica, la asbestosis y algunas conectivopatías23.
La TCAR permite además valorar la presencia de comorbilidades asociadas (enfisema, hipertensión pulmonar [HP], cáncer de pulmón) que pueden condicionar el curso clínico de la enfermedad. Otras enfermedades pulmonares difusas diferentes a las neumonías intersticiales idiopáticas pueden ser también sospechadas mediante la TCAR.
El consenso oficial ATS/ERS/JRS/ALAT 20112 recomienda que el diagnóstico de las neumonías intersticiales idiopáticas se base en el consenso entre el clínico, el radiólogo y el patólogo.
Patrón histopatológicoSi la TCAR no muestra un patrón de certeza típico de NIU, el diagnóstico definitivo deberá realizarse mediante biopsia pulmonar quirúrgica.
Las biopsias se deben obtener en más de un lóbulo, y a ser posible hay que evitar el lóbulo medio y la língula, ya que suelen mostrar cambios inespecíficos que no aportan información diagnóstica. La atelectasia por la extracción se puede minimizar mediante la instilación suave de formol con una aguja, o bien agitando el tejido en el recipiente con formol tras la retirada de la sutura.
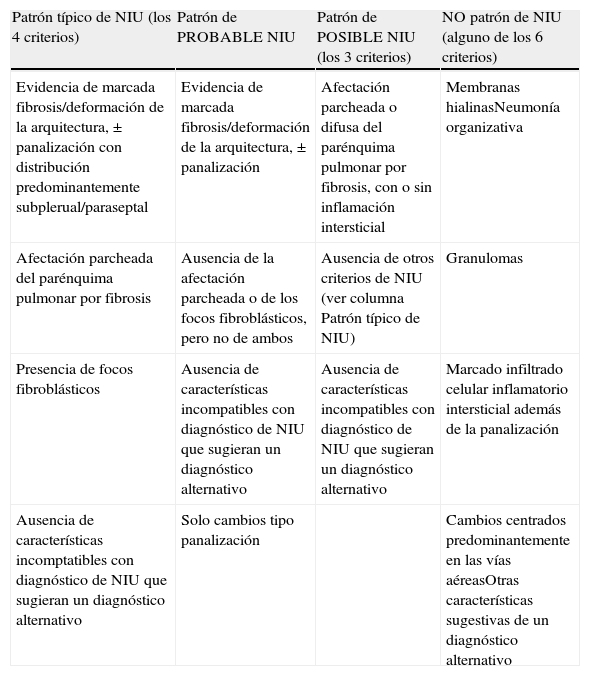
El patrón histológico de NIU (fig. 4) viene definido por el cumplimiento de 4criterios: a)evidencia de fibrosis marcada o distorsión de la arquitectura pulmonar, asociada o no a panalización y con un predominio subpleural y paraseptal; b)presencia de lesiones parcheadas en las que se combinan áreas fibróticas con zonas de pulmón sano; c)presencia de focos fibroblásticos en áreas de interfase de fibrosis con parénquima sano, y d)ausencia de hallazgos histopatológicos inconsistentes con NIU. Entre las características no compatibles con patrón NIU estaría la presencia de membranas hialinas, la presencia de focos con neumonía organizativa, granulomas, marcado infiltrado inflamatorio intersticial alejado de las zonas de panalización, cambios predominantemente centrados en la vía aérea o la presencia de otros hallazgos sugestivos de un diagnóstico alternativo2 (tabla 2).
 con focos de actividad fibroblástica en áreas de interfase (B) y focos de micropanal (C).")
Patrón de neumonía intersticial usual (NIU). Criterios histopatológicos
| Patrón típico de NIU (los 4 criterios) | Patrón de PROBABLE NIU | Patrón de POSIBLE NIU (los 3 criterios) | NO patrón de NIU (alguno de los 6 criterios) |
| Evidencia de marcada fibrosis/deformación de la arquitectura, ± panalización con distribución predominantemente subplerual/paraseptal | Evidencia de marcada fibrosis/deformación de la arquitectura, ± panalización | Afectación parcheada o difusa del parénquima pulmonar por fibrosis, con o sin inflamación intersticial | Membranas hialinasNeumonía organizativa |
| Afectación parcheada del parénquima pulmonar por fibrosis | Ausencia de la afectación parcheada o de los focos fibroblásticos, pero no de ambos | Ausencia de otros criterios de NIU (ver columna Patrón típico de NIU) | Granulomas |
| Presencia de focos fibroblásticos | Ausencia de características incompatibles con diagnóstico de NIU que sugieran un diagnóstico alternativo | Ausencia de características incompatibles con diagnóstico de NIU que sugieran un diagnóstico alternativo | Marcado infiltrado celular inflamatorio intersticial además de la panalización |
| Ausencia de características incomptatibles con diagnóstico de NIU que sugieran un diagnóstico alternativo | Solo cambios tipo panalización | Cambios centrados predominantemente en las vías aéreasOtras características sugestivas de un diagnóstico alternativo |
De Raghu et al.2.
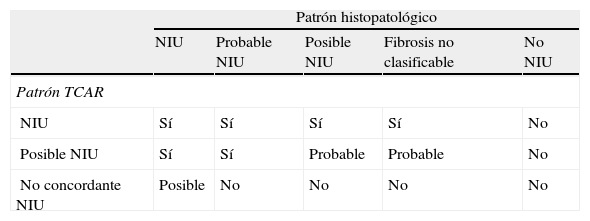
Un patrón histológico indistinguible de la NIU se puede observar en enfermedades sistémicas (p.ej., artritis reumatoide y esclerodermia), neumonitis por hipersensibilidad crónica, neumonitis por fármacos, asbestosis y fibrosis familiares, por lo que hay que descartar la presencia de granulomas, cuerpos de asbesto, infecciones específicas u otros agentes exógenos en la biopsia. Son estas las razones por las que un patrón de NIU no debe ser interpretado directamente como el de FPI sin haber descartado previamente todas estas enfermedades. La integración de los hallazgos de la TCAR con el patrón histopatológico sirve para establecer el diagnóstico de FPI, excluirlo o, si los datos no son concluyentes, mantenerlo como probable o posible (tabla 3).
Integración de los hallazgos de la tomografía axial computarizada de alta resolución (TCAR) con el patrón histopatológico
| Patrón histopatológico | |||||
| NIU | Probable NIU | Posible NIU | Fibrosis no clasificable | No NIU | |
| Patrón TCAR | |||||
| NIU | Sí | Sí | Sí | Sí | No |
| Posible NIU | Sí | Sí | Probable | Probable | No |
| No concordante NIU | Posible | No | No | No | No |
NIU: neumonía intersticial usual.
Modificada de Raghu et al2.
En el momento del diagnóstico, las exploraciones complementarias recomendables son valoración del grado de tos y disnea, TCAR, exploración funcional respiratoria (espirometría forzada, volúmenes pulmonares, capacidad de transferencia pulmonar de monóxido de carbono [DLCO]), gasometría arterial, prueba de la marcha de los 6min (PM6M) y ecocardiograma. En el seguimiento de la evolución, las exploraciones a realizar dependerán del estado del paciente. En los casos con alteraciones leves de la función pulmonar y ausencia de limitación al ejercicio se debe evaluar el grado de tos y disnea y realizar pruebas funcionales respiratorias (capacidad vital forzada [FVC], DLCO), pulsioximetría en reposo, radiografía de tórax (opcional) y PM6M (opcional) cada 3-6meses. No obstante, deben practicarse en períodos más cortos de tiempo si existen signos de progresión de la enfermedad. La medición de la TLC no es precisa para valorar la progresión de la enfermedad. En presencia de progresión de la enfermedad debe también realizarse gasometría, radiografía de tórax y PM6M de forma secuencial, cada 3meses, con el fin de evaluar la oxigenación y la posibilidad de instaurar oxigenoterapia domiciliaria. No es preciso realizar la TCAR de forma sistemática y secuencial durante el seguimiento. Debe realizarse en casos de progresión de la enfermedad y de sospecha de complicaciones2.
Existen varias escalas para valorar la intensidad de la disnea. Las más utilizadas son la Medical Research Council (MRC), la escala de Borg, el índice de disnea inicial y el índice de disnea de transición24,25. El método más útil para valorar la intensidad de la tos es el cuestionario de Leicester26. Existen diversos cuestionarios para valorar la calidad de vida, y los más utilizados son el SF-36, el St George Respiratory Questionnaire y el University of California San Diego Shortness of Breath Questionnaire27.
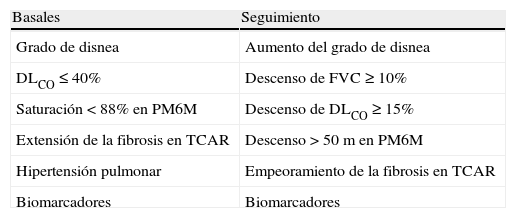
Factores pronósticosLa FPI es una enfermedad de curso clínico variable, por lo que es importante identificar factores que puedan ayudar a definir el pronóstico de los pacientes. Diferentes estudios han evaluado factores clínicos, biomarcadores, parámetros radiológicos y fisiológicos (pruebas funcionales respiratorias y capacidad de ejercicio) y presencia de comorbilidades, asociados con mayor riesgo de mortalidad, tanto en el momento del diagnóstico como evolutivamente durante el seguimiento28. Los factores asociados a peor evolución son (tabla 4):
- -
Edad superior a 70años28.
- -
Comorbilidades asociadas: HP, enfisema y carcinoma broncogénico.
- -
Grado de disnea basal y su incremento en el tiempo. La escala de medición de disnea del MRC ha demostrado ser muy útil para determinar progresión de la enfermedad29.
- -
DLCO menor del 40% (porcentaje del valor predicho) en el momento del diagnóstico30.
- -
Descenso ≥10% de la FVC y ≥15% de la DLCO (porcentaje del valor predicho) en 6-12meses30. El descenso en la FVC es la medida de función pulmonar que mejor predice la mortalidad. Recientemente, Du Bois et al.31 demostraron que los pacientes con una disminución ≥10% de la FVC en 24semanas tienen casi 5veces más riesgo de fallecer en el año siguiente (HR=4,78; IC95%: 3,12-7,33), y aquellos con un descenso del 5-10% de la FVC tienen más del doble de riesgo de mortalidad (HR=2,14; IC95%: 1,43-3,20). Se estima que la diferencia mínima clínicamente importante (DMCI) en la variación de la FVC es del 2 al 6%. En el subgrupo de pacientes con FPI y enfisema asociado, en los que los volúmenes pulmonares son normales o poco disminuidos, la variación de la FVC no predice la supervivencia32.
- -
Desaturación en la PM6M. Tanto la saturación arterial periférica de oxígeno (SpO2)≤88% como la distancia recorrida son predictores de mortalidad en FPI33. La reducción >50m en la distancia recorrida en 24semanas se asocia a un aumento de 4veces el riesgo de muerte en un año (HR=4.27; IC95%: 2,57-7,10). La DMCI se establece en una distancia de 24-45m33. La desaturación discrimina qué parámetro funcional es el más adecuado para el seguimiento. En pacientes con SpO2≤88% la disminución >15% de la DLCO en 6meses es el mejor predictor de mortalidad, mientras que en aquellos con SpO2>88% el parámetro más significativo es la disminución >10% de la FVC34.
También se ha identificado la importancia de la recuperación de la frecuencia cardíaca tras la prueba. El descenso menor de 14latidos tras el primer minuto es un factor independiente de mal pronóstico. La PM6M tiene una buena correlación con el consumo máximo de oxígeno (VO2max) medido en la prueba de ejercicio cardiopulmonar. Un valor de VO2max por debajo de 8,3ml·kg−1·min−1 se asocia a mayor riesgo de mortalidad35.
- -
Extensión de la fibrosis en la TCAR (determinada por la cuantía de la reticulación, panalización y bronquiectasias de tracción) y progresión de la misma36.
- -
Biomarcadores: aunque todavía no suficientemente validados para incorporarlos en el seguimiento de los pacientes, se ha descrito que niveles elevados en el suero de diferentes proteínas asociadas con la patogenia de la FPI pueden predecir mortalidad. Estas proteínas incluyen marcadores de daño de células epiteliales alveolares (KL-6/MUC1, SP-A, MMP-7), de activación de macrófagos alveolares (CCL-18) y de neutrófilos (S100A12, IL-8), y de estrés oxidativo (ICAM-1, VCAM-1)37. Los niveles elevados de fibrocitos en suero se asocian a peor pronóstico y mortalidad precoz. Las concentraciones elevadas de péptido natriurético cerebral (BNP) se relacionan con la presencia de HP y son un predictor de mortalidad en FPI38. En los pacientes en que el diagnóstico de FPI se realiza mediante biopsia pulmonar quirúrgica, la profusión de focos fibroblásticos, cuantificada por métodos semicuantitativos y cuantitativos, también ha demostrado ser un predictor de supervivencia.
Variables predictoras de supervivencia
| Basales | Seguimiento |
| Grado de disnea | Aumento del grado de disnea |
| DLCO≤40% | Descenso de FVC≥10% |
| Saturación<88% en PM6M | Descenso de DLCO≥15% |
| Extensión de la fibrosis en TCAR | Descenso>50 m en PM6M |
| Hipertensión pulmonar | Empeoramiento de la fibrosis en TCAR |
| Biomarcadores | Biomarcadores |
DLCO: capacidad de transferencia pulmonar de monóxido de carbono; FVC: capacidad vital forzada; PM6M: prueba de la marcha de los 6min; TCAR: tomografía axial computarizada de alta resolución.
En la actualidad se están desarrollando escalas multidimensionales para intentar predecir el riesgo individual de mortalidad en pacientes con FPI. Un estudio realizado a partir de datos de 2ensayos clínicos39 ha establecido un modelo clínico compuesto por 4factores (edad, hospitalización, FVC basal y cambio en 24semanas) para la predicción del riesgo de mortalidad a un año. Existen 2estudios recientes: uno retrospectivo40 que emplea el índice GAP (Gender, Age, Physiology), que incluye sexo, edad, FVC y DLCO para predecir el riesgo de mortalidad a uno, 2 y 3años, y otro prospectivo29 que utiliza un sistema de estratificación de riesgo (ROSE) para predicción de supervivencia a 3años, basado en el grado de disnea (escala MRC), la distancia recorrida en la PM6M y el CPI (índice compuesto fisiológico, que incluye FVC, volumen espiratorio máximo en el primer segundo [FEV1] y DLCO). No existe suficiente evidencia científica para el uso de estas escalas en la práctica clínica.
Complicaciones y comorbilidadesLos pacientes con FPI pueden desarrollar complicaciones y comorbilidades que modifican el curso clínico y el pronóstico, bien en relación con mecanismos fisiopatológicos comunes o como enfermedades concurrentes asociadas a la edad.
Exacerbación agudaSe define como el rápido deterioro de la enfermedad en ausencia de infección, insuficiencia cardiaca, embolismo pulmonar u otra causa identificable. El diagnóstico se basa en la combinación de hallazgos clínicos (empeoramiento de la disnea basal en menos de 4semanas) y radiológicos (opacidades en vidrio deslustrado o consolidaciones superpuestas a un patrón típico de NIU en la TCAR)41. Los hallazgos histológicos corresponden habitualmente a patrones de daño alveolar difuso, aunque en ocasiones se caracterizan por neumonía organizativa o focos fibroblásticos añadidos al patrón morfológico de NIU. Su incidencia y su mortalidad varían según las series, dependiendo de los criterios diagnósticos y del tiempo de seguimiento, con incidencia a un año del 9-14% y a 3años del 21-24%38, y mortalidad de hasta el 60-70% en 3-6meses. Varios estudios han establecido posibles factores de riesgo para el desarrollo de exacerbaciones agudas, como la FVC y DLCO bajas y la presencia de enfisema y de HP42.
Hipertensión pulmonarLa HP que aparece en la FPI se incluye en el grupo clínico de HP asociada a enfermedad pulmonar intersticial (grupo3) de la clasificación actualizada de Dana Point y se define por una presión arterial pulmonar media (PaPm) >25mmHg43. Su prevalencia se estima en un 30-45% en pacientes evaluados para trasplante pulmonar, pero es mucho mayor en aquellos con más tiempo de evolución de la enfermedad y en el momento del trasplante. El desarrollo de HP empeora la calidad de vida y la situación funcional de estos pacientes, que presentan más disnea, menor DLCO y capacidad de ejercicio disminuida, con menor distancia recorrida y mayor desaturación en la PM6M43. Además se asocia a una menor supervivencia, con una mortalidad a un año del 28% en pacientes con HP frente al 5,5% en su ausencia44. Aunque en un estudio se han relacionado las resistencias vasculares pulmonares elevadas con mortalidad precoz en FPI45, está por definir qué parámetro hemodinámico tiene más importancia pronóstica en estos pacientes. Los pacientes con FPI pueden presentar otras comorbilidades que también contribuyen al desarrollo de HP, como enfisema, síndrome de apneas-hipoapneas durante el sueño, enfermedad tromboembólica venosa, enfermedad coronaria y disfunción diastólica.
Enfisema pulmonarLa combinación de FPI y enfisema (CFPE) es un síndrome cuyo diagnóstico se basa en los hallazgos en el TCAR de enfisema centrolobulillar y paraseptal en lóbulos superiores y lesiones compatibles con NIU en lóbulos inferiores46. Su prevalencia es del 30 al 47% en pacientes con FPI, y suele aparecer en varones con antecedentes de tabaquismo y disnea importante. La espirometría y los volúmenes pulmonares son normales o están mínimamente disminuidos (efecto opuesto entre la hiperinsuflación debida al enfisema y la pérdida de volumen debida a la fibrosis), pero hay un descenso marcado y desproporcionado de la DLCO e hipoxemia importante con el ejercicio (probablemente por el efecto aditivo del enfisema, la fibrosis y la enfermedad vascular pulmonar). En la CFPE, el descenso de FVC y DLCO es más lento que en los pacientes con FPI aislada y puede llevar a error en las estimaciones pronósticas. Un reciente estudio ha demostrado que en estos pacientes el mejor predictor de mortalidad es el descenso del FEV132.
En la CFPE aparece HP precapilar más frecuente, precoz y marcada que en los pacientes con FPI sin enfisema, que representa un mal pronóstico y es el principal determinante de mortalidad. Está por determinar si la CFPE representa un fenotipo concreto de FPI en fumadores, con factores predisponentes genéticos diferenciados y pronóstico diferente.
Reflujo gastroesofágicoDiversos estudios han documentado la alta prevalencia (66-87%) de RGE en pacientes con FPI, que es asintomático en gran parte de ellos47. La existencia de RGE y hernia de hiato, también más frecuente en pacientes con FPI, puede representar un factor en la patogenia y en su progresión en relación con la presencia de microaspiraciones.
Síndrome de apneas-hipopneas del sueñoLos pacientes con FPI tienen una elevada prevalencia de síndrome de apneas-hipopneas del sueño (SAHS) y otros trastornos respiratorios durante el sueño48. No se ha encontrado correlación entre la gravedad del SAHS y parámetros fisiológicos como FVC, DLCO y volúmenes pulmonares48.
Otras complicacionesExiste mayor riesgo de desarrollar carcinoma broncogénico en los pacientes con FPI. La prevalencia es del 5-10% y aumenta con el tiempo de evolución de la enfermedad y en los pacientes con CFPE49. Además del tabaquismo como factor predisponente, se han descrito diversas mutaciones genéticas que podrían asociarse a su aparición en estos pacientes. En los pacientes con FPI también se encuentra aumentado el riesgo de enfermedades cardiovasculares (enfermedad coronaria y enfermedad tromboembólica venosa)50. El neumotórax puede ser causa de empeoramiento de la FPI. Su incidencia es del 11%2.
TratamientoAntes de iniciar el tratamiento en pacientes diagnosticados de FPI se deben valorar siempre el estadio de la enfermedad, los factores pronósticos y las comorbilidades. El abanico terapéutico incluye51: a)considerar los tratamientos antifibróticos de los que actualmente se dispone; b)evitar las causas agravantes de la enfermedad (RGE, infecciones respiratorias, HP, tabaquismo); c)tratar los síntomas, principalmente la tos y la disnea; d)tener siempre presente el trasplante pulmonar en casos que cumplan criterios, y e)ofrecer terapia paliativa en la fase final de la enfermedad.
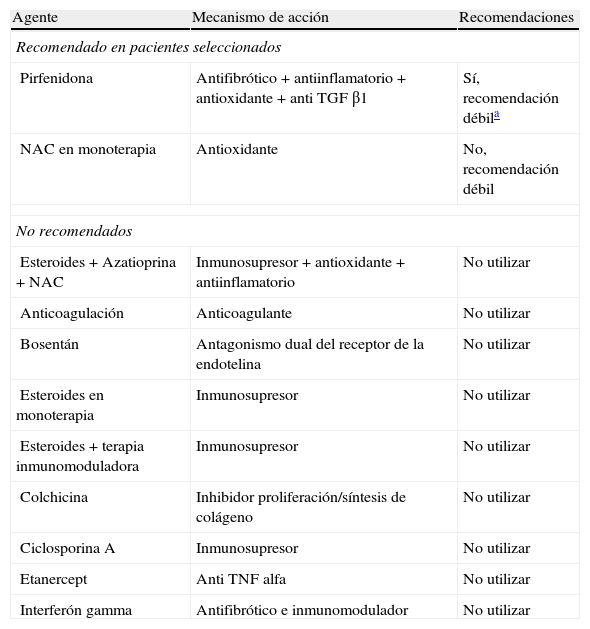
Tratamiento farmacológicoEl enfoque terapéutico en la FPI empezó a cambiar a partir de la nueva hipótesis fisiopatológica de la enfermedad, en la que se planteó el desarrollo del proceso como una alteración reparativa epitelio-mesenquimal que podía iniciarse sin una inflamación previa y donde el tratamiento antiinflamatorio e inmunomodulador no habían demostrado modificar el curso evolutivo de la enfermedad52,53. A partir de este nuevo concepto y tras diversos avances en el conocimiento patogénico de la fibrosis pulmonar se abrieron diferentes vías de investigación con el objetivo de inhibir el proceso fibrogénico desencadenado, lo que fue el inicio de la era «antifibrótica». Dada la ausencia de otras opciones terapéuticas clínicas y a pesar que desde 2003 se constataba la ineficacia de los glucocorticoides en la FPI52,54, hasta el año pasado se seguía considerando en las guías de consenso como opción de tratamiento el uso de glucocorticoides ±inmunomoduladores (azatioprina o ciclofosfamida) ±N-acetilcisteína. En la tabla 5 se especifican las recomendaciones y evidencias para el tratamiento farmacológico de la FPI.
Recomendaciones basadas en la evidencia para el tratamiento farmacológico de la fibrosis pulmonar idiopática (FPI)
| Agente | Mecanismo de acción | Recomendaciones |
| Recomendado en pacientes seleccionados | ||
| Pirfenidona | Antifibrótico+antiinflamatorio+antioxidante+anti TGF β1 | Sí, recomendación débila |
| NAC en monoterapia | Antioxidante | No, recomendación débil |
| No recomendados | ||
| Esteroides+Azatioprina+NAC | Inmunosupresor+antioxidante+antiinflamatorio | No utilizar |
| Anticoagulación | Anticoagulante | No utilizar |
| Bosentán | Antagonismo dual del receptor de la endotelina | No utilizar |
| Esteroides en monoterapia | Inmunosupresor | No utilizar |
| Esteroides+terapia inmunomoduladora | Inmunosupresor | No utilizar |
| Colchicina | Inhibidor proliferación/síntesis de colágeno | No utilizar |
| Ciclosporina A | Inmunosupresor | No utilizar |
| Etanercept | Anti TNF alfa | No utilizar |
| Interferón gamma | Antifibrótico e inmunomodulador | No utilizar |
La N-acetilcisteína (NAC) aumenta la síntesis de glutatión, un potente mediador antioxidante, y disminuye la respuesta fibrótica en modelos animales de fibrosis pulmonar. Un estudio prospectivo multicéntrico en fase iii (estudio IFIGENIA) evaluó la eficacia de la NAC (1.800mg/día) en una cohorte de pacientes con FPI55. Los pacientes recibieron NAC o placebo, en combinación con prednisona y azatioprina. Después de un año, los pacientes que recibieron NAC mostraron una tasa menor de deterioro funcional. El número limitado de pacientes incluidos, la ausencia de grupo placebo y el corto periodo de tiempo evaluado fueron limitaciones muy discutidas. No obstante, la combinación de glucocorticoide, azatioprina y NAC ha sido el tratamiento de elección y recomendado como opción terapéutica en las guías de consenso, hasta que se han conocido los resultados del estudio PANTHER56. El estudio PANTHER comparó la eficacia de placebo vs NAC vs glucocorticoides asociados a NAC y azatioprina. Este estudio ha demostrado mayor mortalidad e ingresos hospitalarios en los pacientes que recibían la triple terapia en comparación con el placebo o con el tratamiento con NAC. Por tanto, no se aconseja utilizar esta triple terapia. En la actualidad el ensayo continúa con solo dos ramas: NAC y placebo. Hasta que se conozcan los resultados no se podrá dilucidar la verdadera eficacia de la NAC como monoterapia en el tratamiento de la FPI.
Pirfenidona (Esbriet®)La pirfenidona es un fármaco con propiedades antiinflamatorias y antifibróticas. Inhibe la proliferación fibroblástica y la síntesis de proteínas pro-fibrogénicas y citocinas. Diversos estudios experimentales han demostrado el efecto antifibrótico de la pirfenidona57. Su eficacia clínica se ha evaluado en 3ensayos clínicos fase iii, multicéntricos, aleatorizados, doble ciego, controlados con placebo en pacientes con FPI en Europa y Estados Unidos (estudios CAPACITY) y Japón. Los resultados de los estudios CAPACITY han mostrado que la pirfenidona a dosis de 2.403mg/24h reduce la progresión de la enfermedad en un 30% y disminuye en un 30% la caída de la FVC. Además, un menor porcentaje de pacientes presentaron progresión rápida (20% pirfenidona respecto a 35% placebo), y se observó una mejoría significativa en la capacidad de esfuerzo, determinada por la distancia recorrida en la PM6M, así como un aumento del intervalo libre de progresión (fig. 5)54,58,59. Hasta la fecha, la pirfenidona es el único fármaco con eficacia contrastada en el tratamiento de la FPI. En marzo de 2011 fue aprobada por la Agencia Europea del Medicamento (EMA) para el tratamiento de la FPI de leve-moderada intensidad, definida por FVC>50% y DLCO>35% (www.ema.europa.eu). En la actualidad, y como consecuencia de los resultados de los ensayos clínicos, se aconseja su uso en pacientes con FVC>50%, y DLCO>35%. Se están desarrollando estudios abiertos con el fin de confirmar la eficacia del fármaco y optimizar sus indicaciones El fármaco está disponible en varios países de la Unión Europea, y próximamente en España. Debe considerarse como el fármaco de primera línea para el tratamiento de la FPI leve-moderada (tabla 5). La dosis que se utiliza es de una cápsula de 267mg/8h durante una semana, en la segunda semana 2cápsulas/8h y, a partir de la tercera semana, 3cápsulas/8h. La duración recomendable del tratamiento es de 12meses como mínimo. Si existe mejoría o estabilización de la enfermedad, parece lógico que debe continuarse el tratamiento. En caso de empeoramiento, debe considerarse en cada paciente si es aconsejable continuar el tratamiento o instaurar otras estrategias terapéuticas. En este contexto, debe puntualizarse que no existen datos que demuestren que la adición de NAC incremente la eficacia de la pirfenidona (fig. 6). Las principales contraindicaciones son hipersensibilidad al fármaco, uso concomitante de fluvoxamina, hepatopatía o nefropatía grave, y embarazo (no comprobado su uso en esta población). La pirfenidona interacciona con el omeprazol, por lo que es aconsejable sustituir este fármaco por pantoprazol. Los efectos secundarios más frecuentes son mareos, fotosensibilidad (imprescindible protección solar cutánea y ocular), molestias digestivas (administrar el fármaco con la ingesta de alimentos) y alteraciones de la función hepática, en general reversibles al disminuir la dosis60.


El nintedanib es un potente inhibidor de la tirosina cinasa que actúa sobre los receptores de factores de crecimiento endotelial vascular (VEGF), plaquetario (PDGF) y fibroblástico (FGF). Los resultados del ensayo clínico en fase ii TOMORROW han evidenciado que la administración de BIBF-1120 150mg/12h ocasiona una tendencia positiva en la reducción de la pérdida de la FVC y de las exacerbaciones de la enfermedad y mejoría de la calidad de vida61. Actualmente se está llevando a cabo un ensayo clínico en fase iii para valorar su eficacia en el tratamiento de la FPI.
Otros fármacos antifibróticosDurante la última década se han realizado varios ensayos clínicos aleatorizados, fase ii y iii, con fármacos considerados experimentalmente anti-fibróticos pero que no han demostrado ninguna eficacia para el tratamiento de la FPI62. Entre ellos se incluyen el mesilato de imatinib (Gleevec), el interferón gamma 1-beta, antagonistas del factor de necrosis tumoral-α (etanercept), anticoagulantes (warfarina), antagonistas de la endotelina (bosentán, macitentán, ambrisentán) y sildenafilo (tabla 5). Por otra parte, están en desarrollo ensayos clínicos en fases i o ii con otras moléculas (www.clinicaltrial.gov): anticuerpo anti-IL13/-IL4 (SAR156597), antagonista del receptor AT1 de la angiotensina-ii (losartán), anticuerpo monoclonal anti-integrina αvβ6 (STX-100), colágeno v (IW-001), anticuerpo anti-IL13 (QAX576), antagonista del factor de crecimiento conectivo (FG-3019), anticuerpo anti-LOXL2 o GS-6624 (AB0024), proteína recombinante pentraxin-2 o rhPTX-2 (PRM-151), y sirolimus. Finalmente, existen infinidad de opciones experimentales consideradas como posibles antifibróticos. Entre ellos cabe destacar el anticuerpo monoclonal anti-TGF-ß1 CAT-192, el factor de crecimiento hepatocítico (HGF), la prostaglandina-E2 (PGE-2) o los ARN de interferencia (nkRNA y PnkRNA). Posiblemente en el futuro la terapéutica antifibrótica eficaz para FPI incluirá la asociación de fármacos que actúen de forma sinérgica sobre diferentes vías patogénicas de la enfermedad.
Tratamiento de las complicaciones y comorbilidadesLas complicaciones de la FPI y la coexistencia de comorbilidades tienen un papel determinante en la evolución de la enfermedad, de ahí la importancia de su detección y tratamiento precoces. Las situaciones más relevantes por su gravedad e implicaciones pronósticas son la exacerbación aguda, la HP y el RGE2.
Exacerbación agudaHasta el momento, no existe ningún ensayo aleatorizado y controlado que sustente un determinado tratamiento para la exacerbación aguda de la FPI, así que las recomendaciones actuales se basan en los resultados de las pocas series publicadas, la mayoría retrospectivas o incluyendo un escaso número de pacientes63. El tratamiento más utilizado son los bolos de corticoides a dosis altas (metilprednisolona 500-1000mg/día) durante 3 días, seguidos de dosis altas de prednisona (0,5mg/kg/día), que se disminuyen de forma paulatina, asociados o no a inmunosupresores como azatioprina, ciclofosfamida o ciclosporina (recomendación débil, calidad de evidencia muy baja). Junto a este tratamiento, el consenso entre expertos defiende el tratamiento de soporte, de forma similar al que se realiza en el síndrome de distrés respiratorio del adulto64 (recomendación débil, calidad de evidencia muy baja). Diversos estudios han demostrado que la ventilación mecánica en pacientes con FPI e insuficiencia respiratoria no es eficaz en la mayoría de los casos65.
Hipertensión pulmonarEl uso de sildenafilo ha sido evaluado en 2ensayos aleatorizados no controlados que muestran mejoría en la capacidad de ejercicio en pacientes con HP asociada a FPI (recomendación débil, calidad de la evidencia muy baja)66,67. Los escasos estudios realizados con epoprostenol o bosentán en pacientes con HP asociada a EPID (solo algunos de ellos tenían FPI) no permiten extraer conclusiones definitivas respecto a la indicación generalizada para su uso en pacientes con HP asociada a FPI. Por tanto, la recomendación actual es que los pacientes con HP moderada-grave constatada por cateterismo derecho (PAPm>35mmHg) pueden ser candidatos a tratamiento con fármacos vasomoduladores como el sildenafilo (recomendación débil, calidad de la evidencia muy baja).
Reflujo gastroesofágicoEl RGE ácido es un factor de riesgo para la aspiración, causa reconocida de neumonitis, por lo que podría contribuir a la inflamación crónica de las vías aéreas y también a la fibrosis. En esta se ha documentado la estabilización clínica una vez tratado el RGE anómalo, bien mediante fármacos o procedimientos quirúrgicos47. Dado el razonable coste y la escasa morbilidad que podría derivarse de los efectos secundarios inducidos por los inhibidores de la bomba de protones, es razonable prescribirlos en pacientes con FPI y RGE ácido demostrado, junto a medidas antirreflujo (recomendación débil, calidad de evidencia muy baja).
Tratamiento no farmacológicoOxigenoterapia domiciliariaLa indicación de oxigenoterapia domiciliaria en pacientes con FPI e hipoxemia en reposo emana, fundamentalmente, de extrapolar las conclusiones de los estudios realizados en pacientes con EPOC e insuficiencia respiratoria crónica. No hay datos concluyentes que avalen el empleo de oxigenoterapia de deambulación para pacientes que desaturan solo durante el esfuerzo, sin insuficiencia respiratoria en reposo68. Dos estudios retrospectivos recientemente publicados apuntan a que la oxigenoterapia domiciliaria podría mejorar la realización de la PM6M en pacientes afectos de EPID69,70 Recientemente, un estudio de cohortes sobre 104pacientes con FPI y 151afectos de otras EPID señala que el mayor requerimiento de oxigenoterapia —es decir, la mayor FiO2 necesaria para mantener una SaO2 basal≥96% antes de realizar la PM6M— es un factor de riesgo independiente asociado a la mortalidad en pacientes con FPI tras un año de seguimiento (recomendación consistente, calidad de evidencia muy baja)71.
Ante la falta de datos concretos en pacientes con FPI, se recomienda administrar oxigenoterapia crónica domiciliaria ante la constatación de hipoxemia significativa en reposo o en la PM6M (SaO2≤88%) (recomendación consistente, calidad de evidencia muy baja).
Trasplante pulmonarEl trasplante pulmonar es el único tratamiento para la FPI en estadios evolucionados que ocasiona una importante mejoría funcional e incremento de la supervivencia a 1, 5 y 10años del 74, el 45 y el 22%, respectivamente72. Estas tasas son significativamente inferiores que las observadas en los pacientes trasplantados por otras enfermedades respiratorias, como el déficit de alfa-1-antitripsina, HP, fibrosis quística o EPOC, pero aunque los resultados del trasplante en pacientes con FPI sean peores, por el momento es la única alternativa terapéutica eficaz, superando todavía los resultados del mejor tratamiento farmacológico disponible. Por ello, los pacientes con FPI con enfermedad progresiva deben evaluarse en una unidad de trasplante pulmonar independientemente del tipo de tratamiento médico que reciban, siempre que no existan contraindicaciones para tal procedimiento quirúrgico (recomendación consistente, calidad de evidencia baja)72.
Rehabilitación respiratoriaLa revisión de The Cochrane Collaboration de 2008 actualizada en 2010 confirma que la rehabilitación es segura en pacientes con EPID (también en el subgrupo de pacientes con FPI) en términos de mejora de la distancia recorrida en la PM6M y de calidad de vida relacionada con la salud73. Sin embargo, no están documentados los efectos beneficiosos de la rehabilitación sobre la supervivencia a largo plazo. Estudios recientes con seguimiento a 6meses muestran que los pacientes con FPI consiguen más beneficios duraderos de los programas de rehabilitación cuando la enfermedad es leve, mientras que el resto de EPID los obtienen independientemente del nivel de gravedad de la enfermedad74,75. Sería recomendable incluir a pacientes con FPI en un programa de rehabilitación respiratoria antes de que la enfermedad alcance estadios evolucionados (recomendación débil, calidad de evidencia baja).
Terapia celular y terapia génicaActualmente, tanto la terapia celular como la terapia génica en la FPI están en fase de estudio experimental, por lo que todavía se deberá esperar un tiempo para conocer su efectividad como tratamiento alternativo o asociado a los tratamientos farmacológicos.
La FPI se caracteriza por la muerte de las células del epitelio alveolar, que son reemplazadas por fibroblastos. Por ello, las aproximaciones terapéuticas basadas en terapias celulares van encaminadas hacia la reposición de las células alveolares encargadas de regenerar el epitelio alveolar. Durante esta última década la implantación de células madre, con la capacidad de proliferar y diferenciarse a células alveolares, se ha planteado como una estrategia terapéutica. Las células madre mesenquimales son probablemente las células más estudiadas y utilizadas, ya que se ha aprovechado su capacidad de diferenciarse a numerosos tipos celulares diferentes. Los resultados obtenidos al administrar células mesenquimales son contradictorios. Por un lado, se ha observado que son capaces de adherirse al alvéolo y adoptar un fenotipo de células alveolares, pero por otro lado también se ha descrito que, en contra de lo esperado, se diferencian a fibroblastos, por lo que estarían aumentando la intensidad de la fibrosis en lugar de disminuirla Esto plantea dudas sobre si la administración de células mesenquimales podría considerarse una terapia ideal en pacientes con FPI76.
Hasta el momento, la mejor opción planteada como terapia celular ha sido la administración de células alveolares tipo ii, que consigue revertir el proceso de fibrogénesis en un modelo animal de fibrosis pulmonar inducida por bleomicina77. Estos resultados indican que la administración de células alveolares tipo ii podría convertirse en una terapia para la FPI. Para ello será necesario esperar los resultados de estudios clínicos actualmente en curso.
Aunque la FPI no es una enfermedad genética, hay numerosos estudios que demuestran que determinados polimorfismos genéticos podrían estar asociados a una mayor susceptibilidad en el desarrollo de FPI8. Al igual que las terapias celulares, la terapia génica solo se ha realizado a nivel experimental, básicamente inhibiendo o administrando diferentes microARN que regulan la expresión de diferentes genes relacionados con la FPI.
Cuidados paliativosEl tratamiento de la tos —especialmente la nocturna que dificulta el sueño— y de la disnea es un factor determinante para mantener una aceptable calidad de vida en pacientes con FPI. La codeína y otros opiáceos y los glucocorticoides a bajas dosis (prednisona 5-10mg/día) han mostrado cierta eficacia en el control de la tos78.
En un ensayo clínico se ha demostrado que la talidomida mejora la tos y la calidad de vida respiratoria (recomendación consistente, calidad de evidencia moderada)79. Dosis bajas de morfina pueden mejorar tanto la sensación de disnea como la tos persistente en pacientes con enfermedad avanzada78.
Los cuidados paliativos deben ir encaminados a mejorar la calidad de vida de los pacientes y sus familias frente a los problemas inherentes a esta enfermedad progresiva y hasta ahora incurable. De esta manera, es importante la identificación, valoración y tratamiento precoces de síntomas como el dolor, la disnea y la tos incoercible, así como cualquier otro síntoma relacionado con la progresión de la enfermedad, tanto en la esfera física como en la psicosocial80. Así, se recomienda que los cuidados paliativos sean considerados parte integrante del tratamiento global de la FPI (recomendación consistente, calidad de evidencia muy baja).
Conflicto de interesesAntoni Xaubet declara haber recibido financiación por impartir conferencias en eventos educacionales y/o por asesoría científica y/o investigación de Intermune, Actelion, Almirall y GSK en relación al tema de la normativa.
Julio Ancochea declara haber recibido financiación por impartir conferencias en eventos educacionales y/o por asesoría científica y/o investigación de Boehringer Ingelheim, Intermune y Zambon en relación al tema de la normativa.
Anna Serrano-Mollar declara haber recibido financiación por impartir conferencias en eventos educacionales y/o por asesoría científica y/o investigación de Intermune en relación al tema de la normativa.







