






Las agudizaciones de la enfermedad pulmonar obstructiva crónica (AEPOC) se caracterizan por una respuesta inflamatoria pulmonar y sistémica, que persiste tiempo después de la resolución clínica. Los mecanismos de este proceso inflamatorio no son bien conocidos.
ObjetivosInvestigar los cambios inflamatorios y sus mecanismos durante las agudizaciones de la EPOC.
MétodosSe determinaron las concentraciones de células inflamatorias en sangre y esputo, óxido nítrico en aire exhalado (FeNO), proteína C reactiva (PCR) en plasma, citocinas (interleucinas [IL] 6, 8, 1β, 10, 12, TNF-α) y SLPI (inhibidor de la leucoproteasa), marcadores de estrés oxidativo, la actividad del factor nuclear kappa B (NF-κB) y de la enzima histona deacetilasa (HDAC) a 17 pacientes durante una AEPOC, en fase estable y a 17 controles fumadores y 11 no fumadores.
ResultadosLas AEPOC se caracterizaron por presentar niveles elevados de FeNO (p<0,05), PCR en plasma (p<0,001) e IL-8, IL-1β, IL-10 en esputo (p<0,05) y mayor activación de NF-κB en macrófagos de esputo en comparación con EPOC estable y controles. Durante la fase estable persisten niveles elevados de estrés oxidativo, SLPI, IL-8, IL-6 y TNF-alfa, sin objetivarse cambios en la actividad HDAC ni en la cantidad de neutrófilos en esputo a pesar de presentar una mejoría significativa (p<0,05) de la función pulmonar.
ConclusionesDurante las AEPOC se observan cambios en marcadores inflamatorios pulmonares y sistémicos que no se resuelven por completo en fase estable. El tratamiento actual no permite modificar la actividad HDAC lo que limita sus efectos antiinflamatorios.
Chronic obstructive pulmonary disease (COPD) is characterised by an inflammatory and systemic response that increases during exacerbations of the disease (ECOPD), although the mechanisms of this inflammatory process are not well known.
ObjectivesTo explore the inflammatory changes and possible mechanisms during ECOPD.
MethodsWe determined the inflammatory cell concentrations in blood and sputum, nitric oxide in exhaled air (FeNO), reactive C-reactive protein (CRP) in plasma, cytokines (IL-6, 8, 1β, 10, 12, TNF-α) and SLPI and total antioxidant activity (TAS) in blood and sputum, the activity of nuclear kappa B factor (NF-kB) and of the histone deacetylase enzymes (HDAC) in 17 patients during ECOPD, in stable phase and in 17 smoking controls and 11 non- smoking.
ResultsECOPD is characterised by higher levels of FeNO (P<.05), plasma CRP (P<.001) and IL-8, IL-1B, IL-10 in sputum (P<.05) compared with stable COPD and controls. The TAS levels in sputum were lower in the exacerbated than in stable phase (P<.05) although significantly higher than the controls (P<.05). These findings were accompanied by a greater activation of NF-kB in sputum macrophages during the ECOPD with no changes in the HDAC activity or in the number of neutrophils in sputum, and a statistically significant deterioration (P<.05) of lung function.
ConclusionsChanges were observed in different pulmonary and systemic inflammatory markers during ECOPD, that were not completely resolved during stability. However, current treatment does not allow the modification of HDAC activity, which limits its anti-inflammatory effects.
La enfermedad pulmonar obstructiva crónica (EPOC) se caracteriza por la presencia de una reacción inflamatoria crónica en respuesta a la exposición prolongada al humo del tabaco1. Los pacientes con EPOC presentan con frecuencia agudizaciones de la enfermedad pulmonar obstructiva crónica (AEPOC). Las AEPOC constituyen un problema socio-sanitario de primera magnitud por su influencia negativa sobre la calidad de vida2,3, la función pulmonar4,5 y el pronóstico de estos enfermos6,7 además de su coste social y económico.
La patogenia de los episodios de AEPOC es poco conocido. Existen diversos estudios que demuestran que los episodios de AEPOC se caracterizan por el aumento de diversos marcadores inflamatorios, incluyendo el número de neutrófilos y macrófagos en la vía aérea y la concentración de citocinas, particularmente IL-6 e IL-88,9, lo que sugiere que el «mecanismo patogénico» fundamental de los episodios de AEPOC es un «brote inflamatorio» pulmonar, con independencia de su causa «desencadenante» (infección, polución atmosférica u otros).
Los mecanismos moleculares de esta respuesta inflamatoria son poco conocidos. La activación del factor nuclear kappa B (NF-κB) juega un papel central en la respuesta inflamatoria en general, y se ha postulado que en la de la EPOC en particular10–12. En condiciones normales, NF-κB está localizado en el citoplasma de la célula y constituido por dos sub-unidades (p65, p50) que son inactivadas por la unión de la proteína I-kB. La fosforilación de la subunidad inhibitoria (I-kB) permite la translocación al núcleo de p65 y p50, su unión a zonas específicas del genoma y la activación de la expresión de diversos genes pro-inflamatorios (TNF-α, IL-8, GM-CSF, iNOS, IL-1β).
En el núcleo, el ADN se encuentra combinado con un grupo de proteínas denominadas histonas13. El nivel de acetilación de estas histonas (medida como actividad histona acetil-transferasa, [HAT]) controla el grado de accesibilidad de diversos factores de transcripción (incluyendo NF-κB) al ADN y, por tanto, controla el nivel de expresión de los genes regulados por dichos factores de trascripción. El mecanismo contrarregulador de esta expresión inflamatoria aumentada consiste en deacetilar dichas histonas nucleares, actividad realizada por las enzimas histona deacetilasas (HDAC). Por ello, una mayor acetilación de las histonas (y por tanto mayor actividad HAT) implica una mayor activación NF-κB y una mayor trascripción de genes inflamatorios. Por el contrario, la deacetilación de las histonas (actividad HDAC) implica que el DNA se hace inaccesible a los factores de trascripción, y por tanto hay menor trascripción inflamatoria. Los glucocorticoides, potentes fármacos antiinflamatorios, inhiben la actividad HAT mediada por NF-κB y reclutan histona deacetilasas (HDAC)14-16 que revierten el proceso de acetilación de histonas e impiden el acceso de factores de trascripción (NF-κB entre otros) al ADN y la trascripción de proteínas inflamatorias. Importantes factores patogénicos en la EPOC, como el estrés oxidativo y el humo del tabaco disminuyen la actividad HDAC de los macrófagos alveolares17-19 y potencian la persistencia del proceso inflamatorio. Se ha sugerido que estos efectos moleculares pueden contribuir a explicar la aparente córtico-resistencia observada en pacientes con EPOC estable20. La alteración de estos mecanismos durante las AEPOC no ha sido explorada.
Diversos estudios sugieren que la resolución de la inflamación puede tardar meses tras el episodio que agudización4. En este estudio pretendemos explorar la hipótesis de que la respuesta inflamatoria tras la agudización es atenuada en parte tras la inhibición de la trascripción inflamatoria dependiente de NF-κB pero no se resuelve por completo debido, al menos en parte, a la persistencia de una actividad HDAC disminuida.
El objetivo de este estudio fue describir los cambios inflamatorios pulmonares y sistémicos durante las agudizaciones de la EPOC y explorar los mecanismos moleculares relacionados con dichos cambios.
MétodosSe reclutaron pacientes con diagnóstico de AEPOC durante las primeras 24 horas de ingreso en el servicio de Urgencias. Estos pacientes tenían diagnóstico de EPOC, definida como un cociente FEV1/FVC < 70% determinado durante estabilidad de acuerdo a los criterios GOLD, una historia de tabaquismo > 15 paquetes/año y diagnóstico de AEPOC al ingreso. Este último realizado por un médico de urgencias, que no participaba ni conocía el estudio, y se basaba en los síntomas del paciente (aumento de disnea, tos y/o cambios en la expectoración) y pruebas complementarias: gasometría arterial, electrocardiograma, radiografía de tórax y analítica sanguínea de rutina.
Del laboratorio de pruebas funcionales respiratorias del servicio de Neumología se reclutaron dos grupos control, uno con historia de tabaquismo mayor a 15 paquetes/año y otro constituido por sujetos no fumadores, todos con un cociente FEV1/FVC > 70%.
Se excluyeron los participantes con historia de asma, bronquiectasias, carcinoma bronquial, neumonía o fallo cardiaco. También se excluyeron aquellos que fueron incapaces de proporcionar una muestra de esputo adecuada o si estaban en tratamiento con teofilina, fármacos antiinflamatorios para el tratamiento de enfermedades inflamatorias como enfermedad de Crohn o artritis reumatoide, o habían recibido tratamiento con antibióticos o glucocorticoides sistémicos durante las últimas 4 semanas. Todos los participantes fueron informados sobre la naturaleza y propósito del estudio y dieron su consentimiento por escrito. El estudio fue aprobado por el comité de Ética de las Islas Baleares.
DiseñoAl ingreso todos los pacientes recibieron tratamiento de acuerdo a las guías internacionales, que incluía broncodilatadores nebulizados (beta-agonistas y anticolinérgicos), glucocorticoides sistémicos (orales o intravenosos) y antibióticos a todos los pacientes que cumplían al menos dos de los siguientes criterios:
- a)
aumento de la disnea habitual del enfermo;
- b)
fiebre;
- c)
aumento del volumen de esputo, y
- d)
aumento del grado de purulencia del esputo21.
Si el paciente aceptaba participar en el estudio, durante las primeras 24 horas de ingreso se realizaba espirometría, gasometría arterial, FeNO y se obtenía una muestra de esputo inducido y sangre.
A los tres meses del alta hospitalaria se programaba una visita ambulatoria en fase estable. Se consideró estabilidad clínica si el paciente no había tenido síntomas de agudización (cambios en la disnea, tos y expectoración) ni necesidad de cambiar su tratamiento habitual. Si el paciente hubiera presentado otro episodio de AEPOC, se citaba tres meses más tarde. Durante la visita ambulatoria se repetían todas las pruebas realizadas durante la hospitalización.
A los controles reclutados en el laboratorio de pruebas funcionales respiratorias, tras aceptar participar en el estudio se les realizó espirometría, gasometría arterial, FeNO y se obtuvo una muestra de esputo inducido y sangre.
Función pulmonarSe realizó espirometría forzada con prueba broncodilatadora (GS, Warren E. Collins, Braintree, MA, EE. UU.) a todos los participantes de acuerdo a las guías internacionales al ingreso y en fase estable22. Los valores de referencia espirométricos usados fueron los de la población mediterránea. Si se disponía de PFR previas se las utilizaba para confirmar el diagnóstico.
Medición del óxido nítrico en aire exhaladoLa medición de óxido nítrico en aire exhalado (FeNO) se realizó con un analizador (Sievers Instruments Inc. Model 280NOA, Boulder, CO, EE. UU.) conectado a un tubo de teflón, siguiendo las recomendaciones del European Respiratory Task Force, continuando con una metodología previamente descrita en nuestro grupo23.
Muestras de esputo y preparaciónLa inducción del esputo y su procesamiento se realizó siguiendo una metodología estandarizada en nuestro laboratorio y previamente descrita24. Brevemente, el esputo obtenido fue incubado con 0.01M DTT en Hank¿s Buffer SALT Solution (HBSS), a 4°C durante 15 minutos, diluido con HBSS, y entonces era filtrado a través de una malla de nylon de 50μm para quitar la mucosidad y detritus, sin remover células y se centrifugaba a 790×g durante 10 minutos. El sobrenadante acelular se extrae y se conserva a -−70°C para posteriores determinaciones. El precipitado celular se resuspende, se realiza recuento celular total con un hemocitómetro de Neubauer, utilizando tinción de azul de tripano para determinar la viabilidad celular. Para obtener el recuento celular se realizan citocentrifugados de la suspensión celular y se realiza tinción con Diff-Quick (International Medical Equipment, San Marcos, CA 92069, EE. UU.). Aquellas muestras con menos de un millón de células se consideraron inadecuadas y fueron excluidas del análisis. Las células se incubaron en una placa de 6 pocillos (multiwell primaria surface modified polystyrene, Falcon 35.3846) a una densidad de 1×106 células/pocillo en 2ml de medio (RPMI 1640, 10% suero fetal bovino, L-glutamina). Tras 4 horas se recoge el sobrenadante y se extraen los macrófagos adheridos que posteriormente son lisados para la extracción de proteínas nucleares (Nuclear extract kit, Active Motif, Carlsbad, CA 92008, EE. UU.).
Determinación de citocinasLa concentración de citocinas (TNF-α, IL-6, and IL-8) se determinó en el sobrenadante por citometría de flujo (CBA, Human inflammation Kit, BD Biosciences, San Jose, CA, EE. UU.) siguiendo las instrucciones del fabricante. La sensibilidad de la prueba, expresada por el fabricante fue la siguiente: 3,6 pg/ml para IL-8, 2,5 pg/ml para IL-6, y 3,7 pg/ml para TNF-α. La determinación de inhibidor de la peptidasa secretoria de los leucocitos (SLPI) se realizó mediante ELISA específico (R&D systems, Minneapolis, MN, EE. UU.).
Actividad antioxidante total (TAS)Se midió la TAS en esputo mediante test colorimétrico (Randox Laboratories Ltd, Crumlin, Reino Unido) utilizando una metodología previamente descrita en nuestro grupo25.
Actividad de enzimas histona deacetilasas (HDAC)Se midió la actividad HDAC en extractos nucleares de macrófagos del esputo mediante una prueba no-isotópica usando un derivado fluorescente derivado de la epsilon-acetil lisina (HDAC fluorescent Activity Assay Kit, BIOMOL, Plymouth, PA, EE. UU.) utilizando las instrucciones del fabricante.
Actividad del factor nuclear kappa B (NF-κB)En extractos nucleares de mácrofagos del esputo de midió la activación NF-κB usando el Kit de ensayo TransAM NF-κB p65 Transcription Factor (Active Motif, Carlsbad, CA, EE. UU.) utilizando las instrucciones del fabricante.
Proteína C reactiva (PCR)La determinación de PCR en plasma se realizó utilizando la técnica de nefelometría siguiendo la técnica estandarizada en el laboratorio central del hospital.
Análisis estadísticoLos resultados se expresaron como media±SEM (o medianas e intervalos en distribuciones no-normales). La actividad HDAC y NF-κB, los valores de IL-8, IL-6, TNF-α, FeNO, TAS, FEV1, SLPI y PCR presentaron distribución normal y se compararon entre los grupos usando ANOVA. Si la ANOVA mostraba diferencias significativas, las comparaciones a posteriori o post hoc (comparaciones múltiples) se realizaron con el test de Tukey-Kramer. Para la comparación entre los grupos de EPOC durante la agudización y la fase estable se utilizó la t Student para muestras pareadas. Se consideró significativo un valor de p<0,05. El análisis se realizó utilizando el software Prisma GraphPad (GraphPad Software Inc., San Diego, CA, EE. UU.).
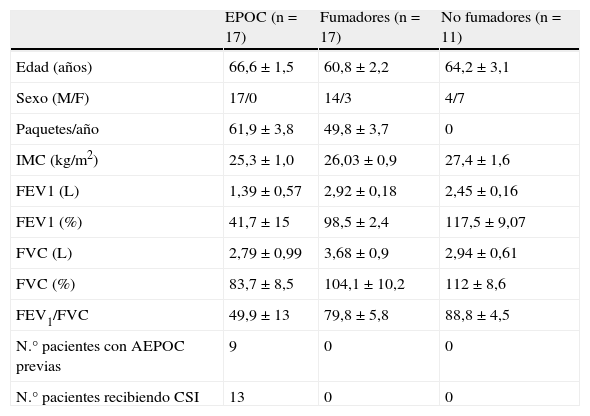
ResultadosDatos clínicosSe reclutaron 17 pacientes con diagnóstico previo de EPOC durante una agudización, que posteriormente fueron re-estudiados en fase estable, y como controles se estudiaron 17 fumadores y 11 no fumadores con función pulmonar normal. Las características demográficas y funcionales de los pacientes se resumen en la tabla 1. Cinco pacientes (29%) con AEPOC tenían un cultivo de esputo con crecimiento de bacterias potencialmente patógenas (BPP): 2 Pseudomonas aeruginosa, 2 Haemophilus influenzae, y 1 Streptococus pneumoniae en el momento del ingreso por agudización y dos pacientes tuvieron crecimiento de Haemophilus influenzae en fase estable. No hubo diferencias en los parámetros analizados a continuación entre los pacientes con cultivo positivo para BPP y los pacientes con cultivo negativo.
Características demográficas y funcionales de los participantes
| EPOC (n=17) | Fumadores (n=17) | No fumadores (n=11) | |
| Edad (años) | 66,6±1,5 | 60,8±2,2 | 64,2±3,1 |
| Sexo (M/F) | 17/0 | 14/3 | 4/7 |
| Paquetes/año | 61,9±3,8 | 49,8±3,7 | 0 |
| IMC (kg/m2) | 25,3±1,0 | 26,03±0,9 | 27,4±1,6 |
| FEV1 (L) | 1,39±0,57 | 2,92±0,18 | 2,45±0,16 |
| FEV1 (%) | 41,7±15 | 98,5±2,4 | 117,5±9,07 |
| FVC (L) | 2,79±0,99 | 3,68±0,9 | 2,94±0,61 |
| FVC (%) | 83,7±8,5 | 104,1±10,2 | 112±8,6 |
| FEV1/FVC | 49,9±13 | 79,8±5,8 | 88,8±4,5 |
| N.° pacientes con AEPOC previas | 9 | 0 | 0 |
| N.° pacientes recibiendo CSI | 13 | 0 | 0 |
Valores expresados como media±desviación estándar.
CSI: corticoesteroides inhalados; FEV1: volumen espiratorio forzado en el primer segundo en fase estable; IMC: índice de masa corporal.
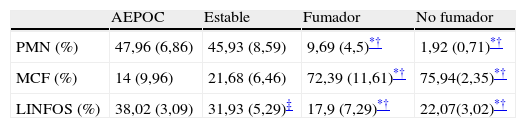
El patrón celular del esputo muestra un predominio neutrofílico en pacientes con EPOC durante la agudización pero que también persiste durante la fase estable, mientras que los controles muestran un predominio de macrófagos y linfocitos (tabla 2).
Patrón celular de esputo
| AEPOC | Estable | Fumador | No fumador | |
| PMN (%) | 47,96 (6,86) | 45,93 (8,59) | 9,69 (4,5)*† | 1,92 (0,71)*† |
| MCF (%) | 14 (9,96) | 21,68 (6,46) | 72,39 (11,61)*† | 75,94(2,35)*† |
| LINFOS (%) | 38,02 (3,09) | 31,93 (5,29)‡ | 17,9 (7,29)*† | 22,07(3,02)*† |
Porcentaje de células expresado como media±desviación estándar.
Los pacientes EPOC agudizados presentaron niveles más elevados de FeNO que se reducen significativamente durante la fase estable (p<0,05), no observándose diferencias entre los individuos en fase estable y controles, tanto fumadores como no fumadores (fig. 1).
 entre pacientes con agudización de EPOC (AEPOC) y fase estable en comparación con controles fumadores y no fumadores.")
Durante las AEPOC también encontramos niveles significativamente elevados de citocinas inflamatorias IL-8, IL-1β, IL-6 y TNF-α en esputo (fig. 2). IL-8 e IL-1β disminuyen significativamente durante la fase estable, pero IL-6 y TNF-α no disminuyen significativamente de la agudización a la fase estable y persisten elevados con respecto a los controles sin EPOC. Los niveles de IL-8 en fase estable fueron significativamente mas elevados que en los controles fumadores y no fumadores. En cambio, los niveles de IL-1β disminuyeron hasta niveles similares a los controles. Las citocinas antiinflamatorias IL-10 y SLPI están aumentadas durante la agudización con respecto a los controles. Los niveles de SLPI permanecen elevados durante la fase estable, pero los niveles de IL-10 disminuyen en fase estable a niveles similares a los controles (fig. 3).
, interleucina 8 (IL-8, Panel B), factor de necrosis tumoral alfa (TNF-α, Panel C) e interleucina 1 beta (IL-1β, Panel D).")
 e interleucina 10 (IL-10, Panel B).")
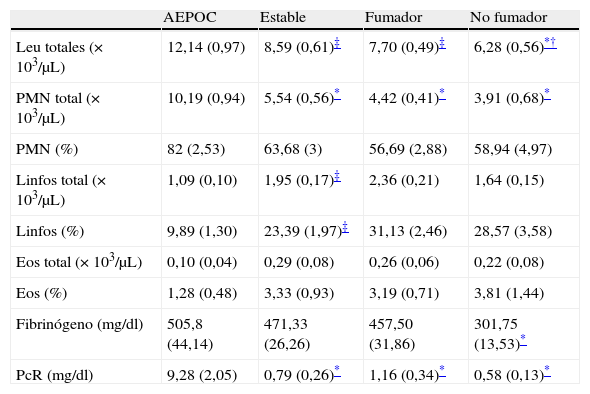
El patrón celular en sangre periférica muestra un significativo aumento de leucocitos polimorfonucleares y disminución de linfocitos en los pacientes agudizados con respecto a los pacientes estables y los controles. Los pacientes con EPOC estable y los fumadores también mostraron mayor número de leucocitos totales que los no fumadores (tabla 3).
Marcadores inflamatorios sistémicos
| AEPOC | Estable | Fumador | No fumador | |
| Leu totales (× 103/μL) | 12,14 (0,97) | 8,59 (0,61)‡ | 7,70 (0,49)‡ | 6,28 (0,56)*† |
| PMN total (× 103/μL) | 10,19 (0,94) | 5,54 (0,56)* | 4,42 (0,41)* | 3,91 (0,68)* |
| PMN (%) | 82 (2,53) | 63,68 (3) | 56,69 (2,88) | 58,94 (4,97) |
| Linfos total (× 103/μL) | 1,09 (0,10) | 1,95 (0,17)‡ | 2,36 (0,21) | 1,64 (0,15) |
| Linfos (%) | 9,89 (1,30) | 23,39 (1,97)‡ | 31,13 (2,46) | 28,57 (3,58) |
| Eos total (× 103/μL) | 0,10 (0,04) | 0,29 (0,08) | 0,26 (0,06) | 0,22 (0,08) |
| Eos (%) | 1,28 (0,48) | 3,33 (0,93) | 3,19 (0,71) | 3,81 (1,44) |
| Fibrinógeno (mg/dl) | 505,8 (44,14) | 471,33 (26,26) | 457,50 (31,86) | 301,75 (13,53)* |
| PcR (mg/dl) | 9,28 (2,05) | 0,79 (0,26)* | 1,16 (0,34)* | 0,58 (0,13)* |
Eos: eosinófilos; linfos: linfocitos; Leu: leucocitos; PMN: leucocitos polimorfonucleares; PcR: proteina C reactiva.
Durante las agudizaciones se encontraron niveles elevados de proteína C reactiva (PCR) y fibrinógeno en plasma en comparación con fase estable y controles fumadores (p<0,001).
No se encontraron diferencias significativas en los niveles séricos de IL-6, 8, 1β, 10, 12, TNF-α medidos en los diferentes grupos.
Mecanismos de inflamaciónLa determinación de indicadores indirectos de estrés oxidativo demuestra una mayor carga oxidativa, traducida por una mayor presencia de antioxidantes en el sobrenadante de esputo de los pacientes con EPOC, tanto estables como agudizados respecto a los controles. Sin embargo, los niveles de TAS en esputo fueron inferiores en agudizados que en fase estable (p<0,05) aunque significativamente mayores en fase estable que los controles (p<0,05) (fig. 4).
. Diferencias en los niveles de activación del factor nuclear kappa B (NF-κB, Panel B) en extractos nucleares de macrófagos de esputo.")
La agudización de EPOC se relacionó con una mayor actividad NF-κB en macrófagos de esputo que disminuye en fase estable (p<0,05) y que fue mayor que los controles no fumadores (p<0,01) (fig. 4). La actividad HDAC en macrófagos estaba disminuida respecto a controles no fumadores (p<0,001) pero sin diferencia entre agudización y fase estable. La disminución de la actividad HDAC se correlacionó de forma significativamente con mayores niveles de IL-8 (r2=0,38, p<0,05) (fig. 5). No se encontraron correlaciones significativas entre los niveles de activación de NF-κB o de estrés oxidativo con el resto de parámetros inflamatorios.
Función pulmonar. Esta reducción esta inversamente correlacionada con los niveles de interleucina 8 en esputo (IL-8, Panel B).")
Los pacientes con EPOC presentaron una recuperación significativa de la función pulmonar a los tres meses de la agudización (p<0,05) (fig. 6) sin encontrarse correlación con los parámetros de inflamación pulmonar. No hubo cambios significativos en la FVC.
Discusión y capacidad vital forzada (FVC) durante la agudización y durante la fase estable.")
En nuestro estudio encontramos que durante las AEPOC se observan cambios en diferentes marcadores inflamatorios pulmonares y sistémicos que se acompañan de un deterioro significativo de la función pulmonar, que podrían estar en relación con un aumento del estrés oxidativo y con una mayor activación del NF-κB. Sin embargo, diversos marcadores de inflamación pulmonar como la concentración de neutrófilos, IL-6 o TNF-α en esputo no cambian significativamente de la agudización a la fase estable, ni tampoco la actividad HDAC, y su afectación se asocia a una mayor producción de IL-8.
Estudios previosLos mecanismos moleculares de la inflamación que ocurren durante las agudizaciones han sido poco estudiados. Algunos trabajos han demostrado una mayor activación del factor de transcripción nuclear NF-κB (mediante localización nuclear por inmunocitoquímica pero sin estudiar su actividad real)26 y el papel del estrés oxidativo durante las agudizaciones27 que daría lugar a una mayor liberación de citocinas que inducen mayor expresión de moléculas de adhesión endotelial como el TNF-α, y quimioquinas que atraen a los neutrófilos en la vía aérea como la IL-8. Estudios previos ya han demostrado un aumento de IL-8 y neutrófilos durante las fases de agudización9. Sin embargo, ningún estudio previo había investigado el mecanismo molecular que regula esta transcripción inflamatoria mediante la activación de la actividad HDAC de las células implicadas en este proceso inflamatorio. Esto puede tener especial relevancia ya que se ha demostrado que incrementar la actividad HDAC durante las agudizaciones potenciando la actividad antiinflamatoria de los glucocorticoides con un estimulador de la actividad HDAC, como es la teofilina a dosis bajas, se traduce en una menor inflamación pulmonar y mejores parámetros clínicos24.
Interpretación de los resultados obtenidosVarios factores se han identificado como desencadenantes de una agudización: infección, polución, etc. Además, hay evidencia suficiente para afirmar que la inflamación juega un rol preponderante en la fisiopatología de la EPOC8,28-30. En nuestro estudio demostramos que, con independencia de la presencia de gérmenes en el esputo de los pacientes agudizados, existe un aumento de la inflamación pulmonar y sistémica. Los pacientes con EPOC estable tienen una respuesta inflamatoria caracterizada por un incremento de macrófagos y linfocitos T CD8+ en la pared del árbol traqueobronquial y de neutrófilos en la luz de las vías respiratorias. Este patrón celular se modifica en las exacerbaciones donde predominan eosinófilos y neutrófilos28. Esta evidencia permite asumir que la inflamación de la vía aérea está amplificada, esto conduce a incremento del tono bronquial, edema de la pared y aumento de la producción de moco, lo que clínicamente se traduce en empeoramiento de la disnea, tos, esputo y alteración del intercambio de gases, dando lugar a los síntomas de la exacerbación. La mayor inflamación de la vía aérea durante las agudizaciones también se acompaña de mayor inflamación a nivel sistémico, lo que se ha sugerido que pudiera tener un papel sobre la mayor morbilidad cardiovascular31. Los mecanismos de este proceso inflamatorio no son bien conocidos, y aquí demostramos que el estrés oxidativo y la activación del factor NF-κB, vía inflamatoria donde convergen diferentes estímulos tanto infecciosos como de otra índole, juegan un papel predominante. Sin embargo, el mecanismo molecular que regula la trascripción de genes inflamatorios en el núcleo celular, que basalmente está disminuida en la EPOC32 y que es responsable de la corticorresistencia observada en el proceso inflamatorio de la EPOC estable33, no se incrementa durante el tratamiento de la agudización con glucocorticoides, lo que creemos que podría ser responsable de la persistencia de marcadores inflamatorios elevados tres meses tras la agudización. En nuestro estudio encontramos que esta actividad se correlaciona con los niveles de IL-8, potente factor quimiotáctico de los neutrófilos, lo que puede contribuir a la persistencia del proceso inflamatorio tras la agudización, y se traduce a su vez en persistencia de neutrófilos en la vía aérea, así como persistencia de niveles elevados de IL-6 y TNF-alfa. Perera et al34 relacionan los cambios inflamatorios presentes en la exacerbación de EPOC y la falta de recuperación y recurrencia en los siguientes 50 días, midiendo las concentraciones de IL-6 y PCR en plasma e IL-6 y 8 en esputo en pacientes EPOC en fase estable, en exacerbación y a los 7, 14 y 35 días post-exacerbación. La falta de mejoría sintomática tras la exacerbación se asoció a elevación persistente de la inflamación sistémica. La persistencia de neutrofilia en esputo y de estrés oxidativo en fase estable observada en nuestro estudio pueden ir en la misma línea. Pinto-Plata et al35 intentaron relacionar citocinas sistémicas y los cambios fisiológicos y clínicos en pacientes hospitalizados por exacerbación de EPOC. Se encontró una correlación significativa entre los cambios en IL-6, IL-8 y LTB4 y los cambios en el FEV1, desde las 48 horas del ingreso hasta 8 semanas tras el alta.
Implicaciones clínicasLa persistencia del proceso inflamatorio tras la exacerbación tiene una traducción clínica relevante. De hecho, es posible que la alta tasa de reingresos en las primeras semanas tras una agudización pueda estar en relación con la persistencia del proceso inflamatorio34. El mecanismo responsable puede tener relación con la falta de activación de la actividad HDAC. Por tanto, fármacos dirigidos a potenciar la actividad HDAC tendrían una traducción clínica relevante ya que al reducir el proceso inflamatorio neutrofílico no sensible al tratamiento corticoideo, podrían reducir la tasa de reingresos y de agudizaciones de estos pacientes. Obviamente, son necesarios ensayos clínicos aleatorizados para confirmar estas implicaciones clínicas propuestas.
Limitaciones potencialesEl objetivo de este estudio fue describir los mecanismos moleculares potenciales que juegan un papel en el proceso inflamatorio subyacente en la EPOC. Se ha estudiado la microbiología de esputo como posible causa de agudización, pero el papel preciso de factores proinflamatorios distintos a la infección bacteriana como la polución ambiental, cambios de temperatura o las infecciones víricas no ha sido determinado. Del mismo modo la proporción de cultivos con BPP es bajo (29%), sin encontrase diferencias con los pacientes con cultivo negativo, lo cual puede sugerir que el mecanismo molecular último es el mismo. La baja proporción de pacientes colonizados en fase estable (12%) no justificaría el efecto observado sobre la falta de activación de la actividad HDAC ni la persistencia de parámetros elevados.
Un número importante de pacientes (76%) estaba recibiendo tratamiento con corticoides inhalados, por lo que el efecto que este tratamiento puede haber tenido sobre los parámetros inflamatorios no se puede determinar. Otra limitación es el número bajo de controles no fumadores analizados. El tamaño muestral no permite hacer asociaciones con parámetros clínicos pero es suficiente para detectar cambios significativos en diversos parámetros inflamatorios.
ConclusionesLas agudizaciones de la EPOC se acompañan de cambios inflamatorios, tanto a nivel pulmonar como sistémico, que no se resuelven por completo tras la agudización. En el mecanismo molecular de los cambios inflamatorios pulmonares participan el estrés oxidativo y el factor nuclear NF-κB. La mayor activación del factor nuclear NF-κB observada durante la agudización disminuye en fase estable en probable relación con el tratamiento glucocorticoideo, no siendo así el mecanismo último de regulación de la trascripción inflamatoria, la actividad HDAC, que podría ser responsable de la persistencia de la inflamación neutrofílica tras la agudización.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores agradecen la colaboración de los doctores del servicio de Urgencias: Dr. Joan Vidal, Dr. Javier García y Dr. Jordi Puiguriguer, por su cooperación durante el reclutamiento, al Dr. Alberto Fuster por su ayuda en la realización del ensayo de TAS y a la Dra. Catalina Crespi por su asistencia con el Cytometric Bead Array.







