

La telangiectasia hemorrágica hereditaria (THH) es una enfermedad autosómica dominante caracterizada por la tríada de epistaxis, telangiectasias y malformaciones vasculares. Las complicaciones vasculares pulmonares asociadas a esta enfermedad incluyen malformaciones arteriovenosas (MAV) pulmonares y, de forma menos frecuente, hipertensión pulmonar (HP).
El presente caso clínico hace referencia a un paciente con múltiples MAV pulmonares e HP en el contexto de posible THH. Se procedió a embolización de una MAV y se inició tratamiento específico de hipertensión arterial pulmonar con un antagonista de receptores de endotelina. A continuación se describe su mejoría funcional y hemodinámica tras 3 años de seguimiento.
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant disorder characterized by the triad of epistaxis, telangiectasia and vascular malformations. Pulmonary vascular complications associated with this disease include pulmonary arteriovenous malformations (AVM) and, less frequently, pulmonary hypertension (PH).
We report the case of a patient who presented multiple pulmonary AVM and PH probably due to HHT. Embolization was carried out on one of the AVM and the patient received specific pulmonary arterial hypertension treatment with an endothelin receptor antagonist. We also described the patient's functional and hemodynamic improvement after almost 3 years of follow-up.
La telangiectasia hemorrágica hereditaria (THH) o síndrome de Rendu-Osler-Weber es un trastorno autosómico dominante con penetrancia variable que se caracteriza por la presencia de epistaxis, telangiectasias y malformaciones vasculares en distintos territorios, tales como el pulmonar, el gastrointestinal, el hepático y el cerebral1. Se trata de una enfermedad poco frecuente, con una prevalencia estimada de 1:5.000 a 1:10.000 casos y con variabilidad regional2.
La presencia de manifestaciones pulmonares vasculares no es despreciable. Se pueden encontrar malformaciones arteriovenosas (MAV) pulmonares hasta en el 45% de los pacientes con THH3. La hipertensión pulmonar (HP) es una complicación menos frecuente y puede ser de 2 tipos: por un lado, HP asociada a malformaciones vasculares hepáticas o pulmonares, generalmente en el contexto de estados hipercinéticos (y asociado frecuentemente a episodios de insuficiencia cardíaca), y por otro, aunque menos frecuente, HP pre-capilar como una forma de hipertensión arterial pulmonar (HAP). El estudio hemodinámico pulmonar es la herramienta diagnóstica para diferenciar ambas situaciones.
A continuación se describe el caso de un paciente con THH e HP.
Caso clínicoPaciente varón de 58 años de edad, ex fumador (con una dosis acumulada de 20 paquetes-año) y ex enolismo moderado, y con antecedentes personales de hipertensión arterial sistémica, linfangiectasias duodenales, epistaxis de repetición y cirrosis hepática enólica ChildA (MELD13) con un único episodio de hemorragia digestiva alta por sangrado de varices esofágicas. Tiene 2 hijas, una de las cuales refería episodios de epistaxis de repetición.
Ingresó en nuestro hospital por clínica de infección respiratoria e insuficiencia respiratoria aguda con una saturación de O2 de 77% (FIO20,21). En la radiografía de tórax se observó un infiltrado en la base derecha que se interpretó como una probable neumonía adquirida en la comunidad y se inició tratamiento antibiótico con fluoroquinolonas (levofloxacino). Ante la persistencia de hipoxemia severa refractaria se realizó un ecocardiograma doppler con contraste que evidenció el paso de burbujas de las cavidades derechas a las izquierdas a partir del quinto latido y signos de HP, estimándose una presión arterial pulmonar sistólica de 81mmHg. La tomografía computarizada de tórax mostró múltiples fístulas arteriovenosas, la mayor de 3cm en el lóbulo inferior derecho, y signos radiológicos de HP.
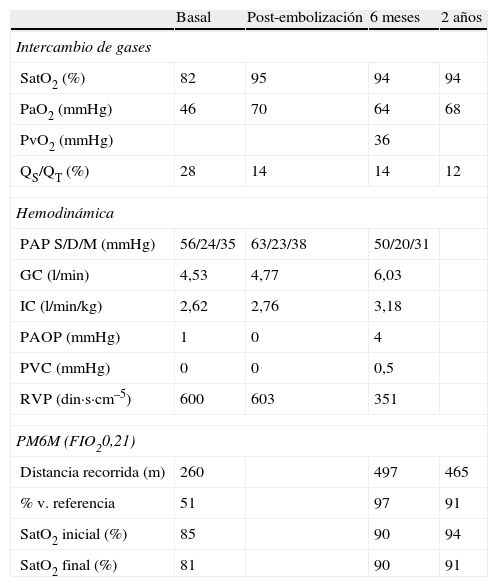
La gammagrafía ventilación-perfusión con macroagregados de albúmina marcada con tecnecio informó de captación homogénea en el parénquima pulmonar y captación extrapulmonar del radiotrazador en ambas siluetas renales, estómago, glándulas salivales y cerebro, sugestivo de cortocircuito sistémico. El cateterismo cardiaco derecho confirmó la presencia de HP con un valor de presión arterial pulmonar media (PAPm) de 42mmHg, gasto cardíaco (GC) ligeramente disminuido (4,53l/min) y aumento de la resistencia vascular pulmonar (RVP) (600din·s·cm–5). La presión venosa central (PVC) y la presión de oclusión arterial pulmonar (PAOP) eran normales (con valores de 0 y 1mmHg, respectivamente). El cálculo de cortocircuito pulmonar (shunt) mediante gasometría arterial basal y con oxígeno al 100% puso de manifiesto un Qs/Qt del 28% (tabla 1). La angiografía pulmonar confirmó la presencia de una MAV de 35mm en el lóbulo inferior derecho, y en el mismo momento se procedió a la embolización de la misma mediante la colocación de un dispositivo intravascular tipo Amplatzer, procedimiento que se realizó sin complicaciones. Las pruebas de imagen de control del dispositivo mostraron la colocación correcta del mismo (fig. 1). Tras el cierre de la MAV, el estudio hemodinámico pulmonar mostró disminución del GC (3,90l/min) y aumento transitorio de la RVP (778din·s·cm−5) que volvieron a sus valores basales al cabo de 8h. La prueba de reactividad vascular pulmonar con óxido nítrico (NO) inhalado a 40ppm no fue significativa. Con el diagnóstico de HP pre-capilar se inició tratamiento con antagonistas de los receptores de endotelina (bosentán a dosis inicial de 62,5mg/12h y a partir de los 28 días a dosis de 125mg/12h).
Caracterización del intercambio de gases, la hemodinámica pulmonar y la prueba de marcha de 6 minutos
| Basal | Post-embolización | 6 meses | 2 años | |
| Intercambio de gases | ||||
| SatO2 (%) | 82 | 95 | 94 | 94 |
| PaO2 (mmHg) | 46 | 70 | 64 | 68 |
| PvO2 (mmHg) | 36 | |||
| QS/QT (%) | 28 | 14 | 14 | 12 |
| Hemodinámica | ||||
| PAP S/D/M (mmHg) | 56/24/35 | 63/23/38 | 50/20/31 | |
| GC (l/min) | 4,53 | 4,77 | 6,03 | |
| IC (l/min/kg) | 2,62 | 2,76 | 3,18 | |
| PAOP (mmHg) | 1 | 0 | 4 | |
| PVC (mmHg) | 0 | 0 | 0,5 | |
| RVP (din·s·cm–5) | 600 | 603 | 351 | |
| PM6M (FIO20,21) | ||||
| Distancia recorrida (m) | 260 | 497 | 465 | |
| % v. referencia | 51 | 97 | 91 | |
| SatO2 inicial (%) | 85 | 90 | 94 | |
| SatO2 final (%) | 81 | 90 | 91 | |
SatO2: saturación de oxígeno por pulsioximetría; PaO2: presión parcial de oxígeno en sangre arterial; PvO2: presión parcial de oxígeno en sangre venosa mixta; QS/QT: cortocircuito pulmonar; PAPS/D/M: presión arterial pulmonar sistólica/diastólica/media; GC: gasto cardíaco; IC: índice cardíaco; PAOP: presión de oclusión arterial pulmonar; PVC: presión venosa central; RVP: resistencia vascular pulmonar; PM6M: prueba de marcha de 6min; FIO2: fracción inspirada de oxígeno.
 En la radiografía de tórax tras el cierre de la malformación arteriovenosa (MAV) pulmonar se observa el dispositivo intravascular Amplatzer (destacado con un círculo blanco). B) La tomografía computarizada de tórax con contraste intravenoso pone de manifiesto la MAV pulmonar ocluida por el dispositivo intravascular Amplatzer (flecha blanca).")
A) En la radiografía de tórax tras el cierre de la malformación arteriovenosa (MAV) pulmonar se observa el dispositivo intravascular Amplatzer (destacado con un círculo blanco). B) La tomografía computarizada de tórax con contraste intravenoso pone de manifiesto la MAV pulmonar ocluida por el dispositivo intravascular Amplatzer (flecha blanca).
El paciente evolucionó de forma favorable, con mejoría de la oxigenación arterial y disminución del cortocircuito pulmonar (QS/QT).
Con los antecedentes familiares descritos, y puesto que la THH tiene carácter hereditario, se procedió al estudio genético. La secuenciación del gen BMPR2 fue normal, y la secuenciación completa del gen ACVRL1 (activin A receptor type II-like 1 [ALK1]) detectó la variación nucleotídica c.355+C>T (rs2071218), que ha sido descrita en la bibliografía como polimorfismo del tipo single nucleotide polymorphism (SNP) sin asociación clínica con el síndrome de Rendu-Osler-Weber. Sin embargo, a pesar de que el paciente no es portador de ningún cambio nucleótido que pueda considerarse como una mutación patogénica asociada al síndrome de Rendu-Osler-Weber, en toda la región codificante del gen ACVRL1, así como en las regiones intrónicas adyacentes, la ausencia de mutaciones en las regiones del gen ACVRL1 analizadas no permite confirmar ni descartar el diagnóstico de THH.
Seis meses después del cierre de la MAV y del inicio del tratamiento específico de HAP, el cateterismo de cavidades derechas mostró mejoría significativa de la hemodinámica pulmonar (tabla 1). En la prueba de marcha de 6min (PM6M) caminó 497m, recorriendo el 97% de la distancia predicha sin presentar disnea ni desaturación.
El paciente ha seguido controles en el servicio de neumología durante los últimos 3años. Actualmente presenta estabilidad en el intercambio gaseoso (persiste con hipoxemia leve, presión parcial de oxígeno en sangre arterial de 68mmHg y presión parcial de monóxido de carbono de 35mmHg, pH7,43) y de la clase funcional (CFI), sin limitación de la capacidad de ejercicio.
DiscusiónLa presencia de HP en los pacientes con THH requiere estudios complejos para determinar a qué tipo de HP corresponde (aislada como HAP o secundaria a insuficiencia cardíaca hipercinética). Nuestro paciente presentaba HP, la cual es clínica e histológicamente indistinguible de la HAP idiopática. El estudio hemodinámico pulmonar mostró, además de aumento de la PAPm, aumento de la RVP y PAOP <15mmHg, pudiéndose descartar la presencia de HP hiperdinámica y/o hipercinética.
La clasificación actual de HAP (Dana Point, 2008) incluye el subgrupo de HAP hereditaria que engloba a los pacientes con HAP idiopática con mutaciones de la línea germinal y los casos de HAP familiar con o sin mutaciones identificadas4. La mayoría de los casos de HAP hereditaria son causados por mutaciones en el gen del receptorII de las proteínas morfogénicas del hueso (BMPR-II)5, un miembro de la superfamilia del receptor del TGF-β. En la HAP asociada a la THH no se encontraron mutaciones del BMPR-II, pero sí en otros genes que codifican componentes del receptor del TGF-β, como el activin receptor-like kinase 1 (ALK-1) y la endoglina (ENG)6,7, localizados en las regiones cromosómicas 12q11-q14 y 9q34.1, respectivamente. En nuestro paciente no detectamos ninguna mutación del BMPR-II pero esto no descarta que no las tuviera, ya que no podemos excluir mutaciones en otras regiones, deleciones o desarreglos genómicos no estudiados. El polimorfismo nucleotídico (SNP) encontrado en el gen ALK1 no ha sido asociado previamente a THH ni a HP.
El hallazgo simultáneo de HAP y MAV pulmonares es poco frecuente y merece especial atención, ya que se ha descrito que la embolización de las MAV puede producir un aumento de la poscarga del ventrículo derecho y desencadenar insuficiencia cardíaca derecha aguda8. Existe poca evidencia sobre los cambios hemodinámicos esperados post embolización. Shovlin et al.9, en un estudio de 143 pacientes con MAV pulmonares que fueron embolizadas no documentaron aumento de las presiones pulmonares, aunque excluyeron los pacientes con HP previa. En nuestro caso, tras la embolización se observó aumento transitorio de la PAPm y de la RVP, con una leve disminución del GC, sin mayores consecuencias.
La experiencia del tratamiento médico de la HAP en la THH está limitada a casos clínicos esporádicos reportados, ya que no existen estudios clínicos aleatorizados, y dada la baja frecuencia de esta patología es poco probable que se puedan desarrollar. Se han utilizado medicaciones aprobadas para el tratamiento de la HAP idiopática con resultados satisfactorios10,11. Nuestro paciente manifestó mejoría de la capacidad funcional (reflejada en la PM6M) y de la hemodinámica pulmonar tras el inicio del tratamiento con bosentán.
En conclusión, la caracterización hemodinámica de la HP en la THH es fundamental para comprender la fisiopatología y determinar el tratamiento. Los casos clínicos reportados de esta patología tan infrecuente y que evalúan los cambios hemodinámicos, los síntomas, la capacidad funcional y la respuesta al tratamiento son útiles ante la ausencia de mayor evidencia científica.
Conflicto de interesesNinguno de los autores tiene conflictos de intereses con el contenido de la nota clínica.







