

Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant disorder characterized by the triad of epistaxis, telangiectasia and vascular malformations. Pulmonary vascular complications associated with this disease include pulmonary arteriovenous malformations (AVM) and, less frequently, pulmonary hypertension (PH).
We report the case of a patient who presented multiple pulmonary AVM and PH probably due to HHT. Embolization was carried out on one of the AVM and the patient received specific pulmonary arterial hypertension treatment with an endothelin receptor antagonist. We also described the patient's functional and hemodynamic improvement after almost 3 years of follow-up.
La telangiectasia hemorrágica hereditaria (THH) es una enfermedad autosómica dominante caracterizada por la tríada de epistaxis, telangiectasias y malformaciones vasculares. Las complicaciones vasculares pulmonares asociadas a esta enfermedad incluyen malformaciones arteriovenosas (MAV) pulmonares y, de forma menos frecuente, hipertensión pulmonar (HP).
El presente caso clínico hace referencia a un paciente con múltiples MAV pulmonares e HP en el contexto de posible THH. Se procedió a embolización de una MAV y se inició tratamiento específico de hipertensión arterial pulmonar con un antagonista de receptores de endotelina. A continuación se describe su mejoría funcional y hemodinámica tras 3 años de seguimiento.
Hereditary hemorrhagic telangiectasia (HHT), or Rendu–Osler–Weber syndrome, is an autosomal dominant disorder with variable penetrance that is characterized by the presence of epistaxis, telangiectasias and vascular malformations in the pulmonary, gastrointestinal, hepatic and cerebral regions.1 It is a rare disease, with an estimated prevalence of 1:5000 to 1:10000 cases, and has regional variability.2
The presence of vascular pulmonary manifestations is significant. Pulmonary arteriovenous malformations (AVM) can be found in up to 45% of patients with HHT.3 Pulmonary hypertension (PH) is a less frequent complication and can be one of two types: PH associated with hepatic or pulmonary vascular malformations, generally in the context of hyperkinetic states (and frequently associated with episodes of heart failure); and, less frequently, pre-capillary PH as a form of pulmonary arterial hypertension (PAH). Pulmonary hemodynamic studies are the diagnostic tool for differentiating between these two situations.
The following is the case report of a patient with HHT and PH.
Case ReportThe patient is a 58-year-old male, ex-smoker (with an accumulated use of 20 pack-years) and ex-moderate drinker, with a personal history of systemic arterial hypertension, duodenal lymphangiectasia, repeated epistaxis and enolic hepatic cirrhosis (Child A – MELD 13) with an episode of upper digestive tract hemorrhage from esophageal varices. He has 2 daughters, one of whom had repetitive episodes of epistaxis.
He was admitted to our hospital due to symptoms of respiratory infection and acute respiratory failure with an O2 saturation of 77% (FIO2 0.21). Chest radiography revealed an infiltrate in the right base that was interpreted as probable community-acquired pneumonia, and antibiotic treatment was initiated with fluoroquinolones (levofloxacin). Given the persistence of severe refractory hypoxemia, Doppler echocardiography with contrast was used, which showed the passage of bubbles from the right cavities to the left after the fifth beat and signs of PH, with an estimated systolic pulmonary artery pressure of 81mmHg. Chest computed tomography showed multiple arteriovenous fistulas, the largest being 3cm in the right lower lobe, and radiological signs of PH.
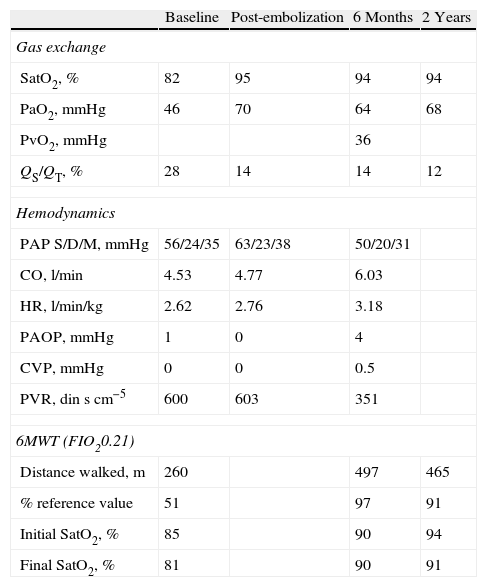
Ventilation–perfusion scintigraphy with macroaggregated albumin marked with technetium reported homogenous uptake in the lung parenchyma and extrapulmonary uptake of the radiotracer in both renal silhouettes, stomach, salivary glands and brain, suggestive of systemic short circuit. Right cardiac catheterism confirmed the presence of PH with a mean pulmonary arterial pressure (PAPm) of 42mmHg, slightly reduced cardiac output (CO) (4.53l/min) and increased pulmonary vascular resistance (PVR) (600dinscm−5). Central venous pressure (CVP) and pulmonary arterial occlusion pressure (PAOP) were normal (with values of 0 and 1mmHg, respectively). The pulmonary shunt equation using baseline arterial blood gases and with oxygen at 100% showed a QS/QT of 28% (Table 1). Pulmonary angiography confirmed the presence of an AVM of 35mm in the right lower lobe; during the procedure, it was embolized with the insertion of an Amplatzer intravascular device, which was done without any complications. Imaging tests showed that the device was correctly inserted (Fig. 1). After the closure of the AVM, the pulmonary hemodynamics study demonstrated a decrease in CO (3.90l/min) and transitory increase of RVP (778dinscm−5), which returned to baseline values after 8h. The pulmonary vascular reactivity test with nitric oxide (NO) inhaled at 40ppm was not significant. With the diagnosis of precapillary PH, treatment was initiated with endothelin receptor antagonist (bosentan at an initial dosage of 62.5mg/12h, and after 28 days at 125mg/12h).
Characteristics of the Gas Exchange, Pulmonary Hemodynamics and 6-min Walk Test.
| Baseline | Post-embolization | 6 Months | 2 Years | |
| Gas exchange | ||||
| SatO2, % | 82 | 95 | 94 | 94 |
| PaO2, mmHg | 46 | 70 | 64 | 68 |
| PvO2, mmHg | 36 | |||
| QS/QT, % | 28 | 14 | 14 | 12 |
| Hemodynamics | ||||
| PAP S/D/M, mmHg | 56/24/35 | 63/23/38 | 50/20/31 | |
| CO, l/min | 4.53 | 4.77 | 6.03 | |
| HR, l/min/kg | 2.62 | 2.76 | 3.18 | |
| PAOP, mmHg | 1 | 0 | 4 | |
| CVP, mmHg | 0 | 0 | 0.5 | |
| PVR, dinscm−5 | 600 | 603 | 351 | |
| 6MWT (FIO20.21) | ||||
| Distance walked, m | 260 | 497 | 465 | |
| % reference value | 51 | 97 | 91 | |
| Initial SatO2, % | 85 | 90 | 94 | |
| Final SatO2, % | 81 | 90 | 91 | |
SatO2, oxygen saturation measured by pulse-oximetry; PaO2, arterial oxygen partial pressure; PvO2, partial pressure of oxygen in mixed venous blood; QS/QT, pulmonary short circuit; PAP S/D/M, pulmonary arterial pressure, systolic/diastolic/mean; CO, cardiac output; HR, heart rate; PAOP, pulmonary artery occlusion pressure; CVP, central venous pressure; PVR, pulmonary vascular resistance; 6MWT, 6-min walk test; FIO2, inspired oxygen fraction.
 Chest radiograph after the closure of the pulmonary arteriovenous malformation (AVM) showing the Amplatzer intravascular device (circled in white); (B) chest computed tomography with intravenous contrast demonstrating pulmonary AVM occluded by the Amplatzer intravascular device (white arrow).")
(A) Chest radiograph after the closure of the pulmonary arteriovenous malformation (AVM) showing the Amplatzer intravascular device (circled in white); (B) chest computed tomography with intravenous contrast demonstrating pulmonary AVM occluded by the Amplatzer intravascular device (white arrow).
The patient evolved favorably, showing improved arterial oxygenation and reduced pulmonary shunt (QS/QT).
With the reported family history, and given the fact that HHT is hereditary in nature, a genetic study was done. BMPR2 gene sequencing was normal, and complete sequencing of the ACVRL1 (activin A receptor type II-like 1 [ALK1]) gene detected nucleotide variation c.355+ C>T (rs2071218), which has been described in the bibliography as a single nucleotide polymorphism (SNP) with no clinical association with Rendu–Osler–Weber syndrome. However, although the patient is not a carrier of any nucleotide changes that could be considered pathogenic mutations associated with the Rendu–Osler–Weber syndrome, not only in the entire codifying region of the ACVRL1 gene, but also in the adjacent intronic regions, the absence of mutations in the regions of the ACVRL1 gene analyzed cannot confirm or rule out the diagnosis of HHT.
Six months after the closure of the AVM and the start of specific PAH treatment, catheterization of the right cavities showed significant improvement in pulmonary hemodynamics (Table 1). During the 6-min walk test (6MWT), the patient walked 497m, which was 97% of the predicted distance walked without presenting dyspnea or desaturation.
The patient has been followed-up in the pulmonology unit for the last 3 years. He currently presents stable gas exchange (mild hypoxemia, arterial oxygen partial pressure of 68mmHg and carbon monoxide partial pressure of 35mmHg, pH 7.43) and functional class I, without any limitations in exercise capacity.
DiscussionThe presence of PH in patients with HHT requires complex studies to determine which type of PH is present (isolated as PAH or secondary to hyperkinetic heart syndrome). Our patient presented PH, which is clinically and histologically indistinguishable from idiopathic PAH. The pulmonary hemodynamic study showed, in addition to increased PAPm, elevated PVR and PAOP <15mmHg, ruling out the presence of hyperdynamic and/or hyperkinetic PH.
The current classification of PAH (Dana Point, 2008) includes the subgroup of hereditary PAH that encompasses patients with idiopathic PAH with germline mutations and cases of family PAH with or without identified mutations.4 The majority of hereditary PAH cases are caused by mutations in the bone morphogenetic protein receptor type II (BMPR-II),5 a member of the superfamily of the TGF-β receptor. In PAH associated with HHT, no BMPR-II mutations were found, although they were found in other genes that code for components of the TGF-β receptor, like activin receptor-like kinase 1 (ALK-1) and endoglin (ENG),6,7 located in the 12q11-q14 and 9q34.1 chromosomal regions, respectively. In our patient, we detected no BMPR-II mutations, but this does not rule them out because we cannot exclude possible mutations in other regions, deletions or genetic disorders that were not studied. The single-nucleotide polymorphism (SNP) found in the ALK1 gene has not been previously associated with HHT or PH.
The simultaneous finding of PAH and pulmonary AVM is uncommon and merits special attention, since it has been reported that embolization of AVM can cause an increase in the postload on the right ventricle and trigger acute right cardiac failure.8 There is little evidence about the hemodynamic changes that are expected post-embolization. Shovlin et al.,9 in a study of 143 patients with pulmonary AVM who were embolized, did not document increased pulmonary pressures, although they excluded patients with previous PH. In our case, after embolization we observed a transitory increase in PAPm and PVR, with a slight decrease in CO, without major consequences.
The experience of medical treatment of PAH in HHT is limited to sporadic reports of clinical cases because there are no randomized clinical studies, and given the infrequency of this pathology it is unlikely that they could be developed. Medications approved for the treatment of idiopathic PAH have been used with satisfactory results.10,11 Our patient showed improved functional capacity (as seen in the 6MWT) and pulmonary hemodynamics after treatment was initiated with bosentan.
In conclusion, the hemodynamic characterization of PH in HHT is fundamental for understanding its physiopathology and for determining treatment. Clinical case reports of this rare pathology that evaluate hemodynamic changes, symptoms, functional capacity and response to treatment are useful given the current lack of major scientific evidence.
Conflict of InterestsNone of the authors have any conflicts of interest regarding the content of this case report.
Please cite this article as: Raimondi A, et al. Hipertensión arterial pulmonar en un paciente con telangiectasia hemorrágica hereditaria. Arch Bronconeumol. 2013;49:119–21.







