


Introducción
El síndrome de apneas-hipopneas del sueño (SAHS) se asocia a un aumento del riesgo de padecer enfermedad cardiovascular1-4. Las bases patogénicas de esta asociación no se conocen, pero uno de los posibles mecanismos implicados es el estrés oxidativo5. Se ha apuntado que la hipoxia intermitente y los episodios de hipoxia-reoxigenación que acompañan a las apneas podrían inducir un aumento de la liberación vascular de radicales libres, favorecer el proceso de formación de la placa de ateroma y aumentar el riesgo cardiovascular en estos pacientes6,7. En esta revisión se exponen algunos conceptos generales sobre la génesis del estrés oxidativo en la pared vascular y la evidencia existente sobre la implicación de este proceso en la patogenia de la enfermedad cardiovascular de los pacientes con SAHS.
Estrés oxidativo
Generalidades
Aunque los radicales libres o especies reactivas de oxígeno (RLO) tienen unas funciones fisiológicas bien definidas (p. ej., la generación de descargas oxidativas por parte de los neutrófilos o inducir la activación de vías de señalización intracelulares relacionadas con el crecimiento), son moléculas altamente reactivas que pueden dar lugar a daño celular8. La producción incontrolada de RLO puede ser fuente de enfermedad a través de la alteración de macromoléculas (lípidos, proteínas, hidratos de carbono y ácidos nucleicos) y diversos procesos celulares (funcionalidad de las membranas, producción de enzimas, inducción génica). Los RLO se producen durante las reacciones metabólicas, cuando las células del organismo transforman los alimentos en energía especialmente en situaciones de hiperoxia, ejercicio intenso e isquemia. También se producen por la exposición a determinados agentes externos como las radiaciones ionizantes, luz ultravioleta o humo del tabaco. Los RLO inorgánicos más importantes son el oxígeno molécular (O2), el radical anión superóxido (O2-), el radical hidroxilo (HO-) y su precursor inmediato el peróxido de hidrógeno (H2O2); de los orgánicos, el radical peroxilo (ROO-), el hidroperóxido orgánico (ROOH) y los lípidos peroxidados8-10.
El organismo posee unos sistemas antioxidantes de defensa para la eliminación inmediata de los radicales libres. Además existen sustancias antioxidantes que el organismo no puede sintetizar y que adquirimos por medio de la dieta (tabla I). Se define como sustancia antioxidante cualquier sustancia que, cuando está presente a concentraciones bajas en relación con el sustrato oxidable, retrasa o previene significativamente la oxidación de dicho sustrato, en el que se incluyen las moléculas orgánicas e inorgánicas que se encuentran en las células vivas, como proteínas, lípidos, hidratos de carbono o el ADN.

Si el equilibrio entre los radicales libres y los sistemas antioxidantes se rompe a favor de los primeros, se produce el denominado estrés oxidativo, que desempeña un importante papel en numerosos procesos degenerativos como el envejecimiento, la arteriosclerosis o el cáncer8-10. Los RLO cumplen un papel importante en el desarrollo de vasculopatías, entre ellas la arteriosclerosis, la hipertensión y la reestenosis postangioplastia11. Actualmente está claro que muchos RLO se producen en la pared arterial y que, solos o en combinación, contribuyen a diferentes anormalidades asociadas con la enfermedad vascular12,13.
Especies reactivas de oxígeno y pared vascular
Existen diversos RLO que desempeñan un papel importante en la fisiología y fisiopatología vasculares. Los más importantes son el óxido nítrico (NO), el O2-, el H2O2 y el peroxinitrito (ONOO-)12. Los RLO están implicados en algunas funciones fundamentales de la pared arterial. El NO es un mediador crucial de la vasodilatación dependiente del endotelio , mientras que el O2 y H2O2 intervienen en el crecimiento, diferenciación y apoptosis de las células musculares lisas14,15. Por otra parte, la peroxidación lipídica y nitración proteica inducidas por el ONOO- son acontecimientos aterogénicos tempranos16. Cada uno de los RLO deriva de reacciones químicas o enzimáticas específicas (fig. 1). El NO se produce en las células endoteliales por la activación de la enzima NO sintetasa endotelial (eNOS), pero los macrófagos y células musculares lisas pueden expresar NO sintetasa inducible y contribuir a la producción de NO.
El NO es un mediador crucial de la vasodilatación dependiente del endotelio. También participa en la agregación plaquetaria y en el mantenimiento del equilibrio entre el crecimiento y la diferenciación de las células musculares lisas. Diversas hormonas vasodilatadoras y fuerzas físicas pueden activar la enzima eNOS. La expresión de NO sintetasa inducible en macrófagos y células musculares lisas origina una elevación de las concentraciones de citocinas que da lugar a una respuesta inflamatoria local. Además, en determinadas circunstancias la eNOS está desacoplada por déficit de un cofactor esencial, la tetrahidrobiopterina, y se produce O2- en vez de NO. Es decir, las enzimas NO sintetasa son potenciales fuentes de NO y O2- dependiendo de las circunstancias ambientales13,17,18.
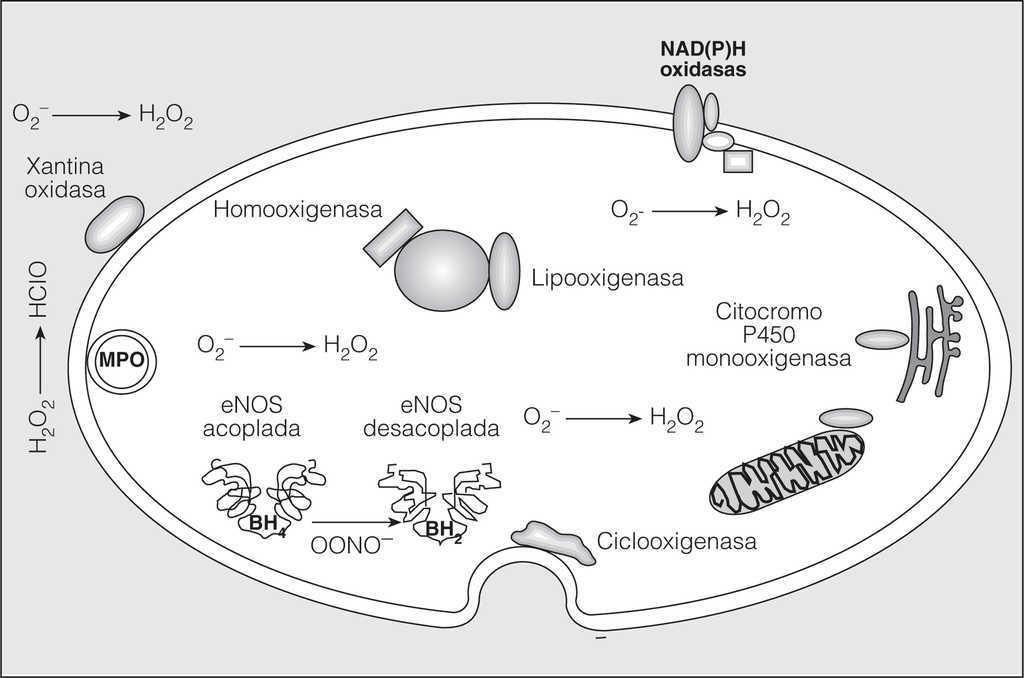
Fig. 1. Fuentes potenciales de radicales libres de oxígeno. Muchas enzimas, como las que participan en el transporte mitocondrial de electrones, xantina oxidasa, ciclooxigenasas, lipooxigenasas, mieloperoxidasas, citocromo P450 monooxigenasa, óxido nítrico sintetasa (eNOS) desacoplada, hemooxigenasas, peroxidasas y dinucleótido de nicotinamida y adenina o fosfato de dinucleótido de nicotinamida y adenina --NAD(P)H oxidasas--, producen radicales libres de oxígeno. Dependiendo de su localización en la célula, estos radicales libres de oxígeno se generan intracelular, extracelularmente o en compartimientos intracelulares específicos. O2-: radical anión superóxido; H2O2: peróxido de hidrógeno; OONO-: peroxinitrito; BH4: tetrahidrobiopterina; BH2: dihidrobiopterina. (Adaptada con permiso de Griendling et al12.)
Todas las células vasculares producen O2- y H2O2. El O2- es el resultado de la reducción de un electrón del oxígeno por una variedad de oxidasas. Cuando el O2 se produce en concierto con NO, ambos reaccionan rápidamente para dar lugar a la molécula altamente reactiva ONOO-. El ONOO- es un importante mediador de la oxidación lipídica, como la oxidación de lipoproteínas de baja densidad (LDL), con importantes efectos proaterogénicos.
En ausencia de NO, el O2- es rápidamente dismutado hacia un RLO más estable, H2O2, por la superóxido dismutasa, para luego ser convertido en H2O por la catalasa o glutatión peroxidasa12,13. Los efectos del O2- y H2O2 en la función vascular dependen de la cantidad producida. Cuando se forman intracelularmente en pequeñas cantidades, pueden actuar como segundos mensajeros modulando la función de mecanismos bioquímicos que participan en procesos como el crecimiento de células musculares lisas o fibroblastos. Una elevada producción de RLO puede causar lesiones en el ADN, toxicidad celular y apoptosis, como se ha demostrado en células endoteliales y células musculares lisas12. Además de la fuente mitocondrial de RLO, diversas enzimas pueden sintetizar O2- y H2O2, como la dinucleótido de nicotinamida y adenina (NADH) o fosfato de dinucleótido de nicotinamida y adenina (NADPH) oxidasas, cuya actividad puede verse modulada por diversos estímulos. Así, la angiotensina II, el factor de necrosis tumoral alfa, la trombina y el factor de crecimiento derivado de las plaquetas incrementan la actividad oxidasa y elevan las concentraciones de O2- y H2O2 en las células musculares lisas. Fuerzas físicas como los cambios de flujo son también potentes activadores de la producción de O2- en las células endoteliales12,18. Los macrófagos son la principal fuente vascular de O2- en situaciones patológicas. Oxidan las LDL a través de la activación de diversas enzimas. Los neutrófilos y monocitos pueden también secretar mieloperoxidasa, la cual es importante en el inicio de la peroxidación lipídica12,16.
Los macrófagos también liberan al espacio extracelular RLO que pueden activar metaloproteinasas. Una vez activadas, las metaloproteinasas pueden degradar la matriz extracelular, debilitar la capa fibrosa de la placa de ateroma y facilitar su rotura16. Además, productos más estables de los RLO pueden influir en la función celular a través de sus acciones sobre mecanismos específicos, o bien actuando como ligandos para receptores nucleares y de membrana. Los RLO pueden modificar directamente la afinidad de ciertos factores de transcripción (factor nuclear kB o proteína activadora 1) para sus sitios de unión en el ADN. Los RLO regulan diversas clases de genes, incluyendo moléculas de adhesión, factores quimiotácticos, enzimas antioxidantes y sustancias vasoactivas. Algunas de estas acciones son claramente una respuesta adaptativa como, por ejemplo, la inducción de las enzimas superóxido dismutasa y catalasa por H2O2. La sobrerregulación de moléculas de adhesión y quimiotácticas por mecanismos sensibles a oxidantes es de particular relevancia en las enfermedades vasculares. Estas moléculas promueven la adhesión y migración de monocitos hacia el interior de la pared. Por otra parte, el NO inhibe la inducción transcripcional de moléculas de adhesión por citocinas. En conjunto estos mecanismos combinan la supresión de la expresión de moléculas de adhesión en la pared arterial normal y la inducción de su expresión en vasculopatías12,19.
Asimismo, no puede descartarse la influencia de factores genéticos que modulen la generación de RLO en el tejido vascular. En este sentido, se ha identificado un polimorfismo en el gen CYBA (C242T), que codifica la subunidad p22phox de la enzima NADPH oxidasa y que se ha relacionado con la actividad de ésta y con la generación de O2- en el tejido vascular20.
Monitorización de la formación de radicales libres de oxígeno
Los RLO son moléculas evanescentes y, como consecuencia, su medición es compleja y ha experimentado diversos cambios. Los índices utilizados tradicionalmente, como la medida de la susceptibilidad de la LDL a la oxidación, han dado paso al desarrollo de diversos biomarcadores de estrés oxidativo. Se trata de identificar compuestos estables derivados de la acción de RLO como productos de la peroxidación lipídica (como los isoprostanos), proteínas modificadas (fibrinógeno nitratado) o índices de ADN modificado (8-oxodeoxiguanosina). También se han desarrollado anticuerpos dirigidos contra epítopos presentes en las LDL oxidadas que pueden utilizarse para medir la peroxidación lipídica. La espectrometría de masas se ha empleado asimismo para detectar aminoácidos que han sufrido modificación oxidativa.
La liberación excesiva de RLO también puede evaluarse a partir de diversos marcadores indirectos, como son la detección de una activación de genes sensibles a RLO, la reducción de la liberación de NO, la elevación de las concentraciones de homocisteína o la reducción de los valores de antioxidantes como enzimas y/o vitaminas. Estos métodos continúan perfeccionándose y probablemente surjan nuevos marcadores que nos permitirán medir la acción de los RLO de una manera más exacta y precisa21-24.
Estudios en humanos
Los más recientes marcadores de estrés oxidativo indican que éste es una característica presente en diversas enfermedades cardiovasculares y se detecta en pacientes con distintos factores de riesgo cardiovascular. En pacientes con hipercolesterolemia se han encontrado concentraciones elevadas de isoprostanos, así como anticuerpos anti-LDL oxidada. El tratamiento con estatinas parece reducir las concentraciones de estos marcadores25,26. En fumadores también se han detectado valores elevados de dichos marcadores, así como un incremento en la modificación oxidativa del fibrinógeno y concentraciones urinarias elevadas de 8-oxodeoxiguanosina27,28. Asimismo, se han relacionado con la obesidad las concentraciones elevadas de isoprostanos, que disminuyen tras el adelgazamiento29.
Hay una gran cantidad de estudios experimentales que evidencian los efectos beneficiosos de los antioxidantes. Por ejemplo, la vitamina C reduce la adhesión de los monocitos a las células endoteliales, inhibe la oxidación de LDL y estimula la actividad de eNOS. La vitamina E también inhibe in vitro la adhesión leucocitaria y la oxidación de LDL30-33. Numerosos estudios clínicos han intentado validar estas observaciones en humanos. El tratamiento con vitamina C reduce las concentracions de isoprostanos en individuos fumadores y el efecto beneficioso de esta vitamina sobre la disfunción endotelial está demostrado en pacientes con enfermedad cardiovascular28,34. Los supuestos beneficios de la vitamina E han sido más difíciles de demostrar y no existe acuerdo sobre los efectos de la suplementación con esta vitamina35,36. Aunque existen estudios en los que se ha detectado una mejoría en la función endotelial y en marcadores de oxidación lipídica, así como una reducción de episodios cardiovasculares tras la administración de vitamina E, los resultados de otros estudios de intervención no apoyan estas conclusiones sobre la eficacia de esta vitamina35,36.
Estrés oxidativo y síndrome de apneas-hipopneas del sueño
El papel potencial del estrés oxidativo en la patogenia de las enfermedades cardiovasculares de los pacientes con SAHS deriva de las observaciones que implican, por una parte, la elevada producción de radicales libres en situaciones de hipoxia-reoxigenación y, por otra, la predisposición al desarrollo de arteriosclerosis en estos pacientes6,37. Actualmente disponemos de datos que apoyan esta hipótesis y que se basan en la detección de marcadores directos de estrés oxidativo como son la elevada producción de RLO en leucocitos de pacientes con SAHS o bien en la detección de marcadores indirectos como, por ejemplo, de moléculas derivadas de la activación de genes sensibles a RLO5.
Incremento en la producción de radicales libres de oxígeno en leucocitos
En situaciones de hipoxia o mediante exposición a citocinas u otros factores se produce una activación de los leucocitos que da lugar a una elevada producción de RLO. En 2 estudios se ha observado, tanto tras estimulación como en condiciones basales, un aumento de la producción de RLO en leucocitos aislados de pacientes con SAHS, lo cual indicaría una producción basal constante de RLO en estos pacientes. El tratamiento con presión positiva continua de la vía aérea (CPAP) se acompañó de una reducción de la liberación de RLO en los 2 estudios38,39.
Oxidación de lipoproteínas
Diversas líneas de evidencia mantienen la hipótesis de que la modificación oxidativa de las LDL cumple un papel crucial en la patogenia de la arteriosclerosis. Las LDL oxidadas alteran la respuesta vasomotora e inducen inflamación, ya que promueven la expresión de moléculas de adhesión en el endotelio y la liberación de citocinas y factores de crecimiento. Las LDL oxidadas son captadas ávidamente por los macrófagos, que se transforman en células espumosas. Además, son a la vez consecuencia y mediadores de estrés oxidativo, lo que da lugar a una perpetuación de la respuesta inflamatoria y el daño oxidativo. Se cree que uno de los principales efectos beneficiosos del tratamiento con estatinas consiste en la disrupción del ciclo inflamación-oxidación independientemente de su capacidad para reducir las concentraciones plasmáticas de lípidos16,17,40. Respecto al SAHS, aunque se publicó un estudio donde no se encontraron diferencias entre la susceptibilidad a la oxidación de LDL aisladas del plasma de pacientes con SAHS respecto a individuos sanos41, otros 3 estudios han demostrado un aumento en la oxidación de estas lipoproteínas en pacientes con SAHS y un efecto beneficios o del tratamiento con CPAP sobre dicha oxidación42-44. Estos resultados son indicativos de la importancia de este mecanismo patogénico en relación con el riesgo aterogénico atribuible al SAHS.
Homocisteína
La homocisteína es un aminoácido sulfurado altamente reactivo que causa disfunción endotelial a través de varios mecanismos, como son un aumento de la producción de RLO, menor liberación de NO y alteraciones en la expresión de diversos genes en las células endoteliales45. Se ha demostrado una relación entre las concentraciones elevadas de homocisteína y la frecuencia de enfermedad cardiovascular45,46. También se ha documentado un efecto beneficioso sobre la disfunción endotelial al disminuir los títulos de homocisteína mediante tratamiento con ácido fólico y vitaminas B6 y B1245,46. Sin embargo, no se conoce el efecto que estos tratamientos puedieran tener sobre la reducción de la mortalidad cardiovascular y actualmente están en marcha estudios aleatorizados con el objetivo de conocer el alcance de estos tratamientos.
En un estudio realizado por Lavie et al47 se encontraron concentraciones elevadas de homocisteína en pacientes con SAHS y enfermedad coronaria respecto a pacientes con SAHS e hipertensión y SAHS sin enfermedad cardiovascular. Esta diferencia se detectó también respecto a un grupo de individuos con enfermedad coronaria sin SAHS. No se encontraron diferencias en cuanto a otros factores de riesgo y concentraciones de vitaminas B6, B12 y ácido fólico relacionadas con el metabolismo de la homocisteína. Estos resultados indicarían que la homocisteína puede estar implicada en la patogenia de la enfermedad cardiovascular en pacientes con SAHS.
Oxido nítrico
El NO producido por la enzima eNOS en el endotelio modula el flujo vascular y tiene importantes efectos antiaterogénicos en las plaquetas, células musculares lisas y células endoteliales. En humanos, la función endotelial mediada por NO es deficiente en estados prearterioscleróticos, y diversos estudios consideran que la disfunción endotelial es un predictor independiente de futuros episodios cardiovasculares13. Los mecanismos por los cuales se produce una menor disponibilidad de NO son múltiples. Por una parte, la producción de RLO rápidamente reacciona con el NO y contribuye a su déficit. Además, el ONOO formado por la reacción NO-superóxido tiene efectos nocivos sobre la función vascular a través de la oxidación de proteínas, lípidos y toxicidad celular. Por otra parte, la eNOS puede generar superóxido en vez de NO en determinadas circunstancias, como cuando existe un déficit del cofactor tetrahidrobiopterina o en respuesta a estímulos aterogénicos como la hiperglucemia o la hipercolesterolemia13,17. Diversos estudios han demostrado una mejoría de la función endotelial tras la suplementación con tetrahidrobiopterina48,49.
En varios estudios se han medido, en pacientes con SAHS, los productos estables derivados del NO y se han encontrado concentraciones circulantes disminuidas que aumentaban significativamente tras tratamiento con CPAP50,51. También se han detectado cifras elevadas de dimetilarginina asimétrica, un inhibidor de la síntesis de NO, lo que indicaría que en los pacientes con SAHS diversos mecanismos podrían contribuir a una menor disponibilidad de NO y a una mayor susceptibilidad a la disfunción endotelial52.
Xantina oxidasa
La xantina oxidasa cataliza la degradación de hipoxantina a ácido úrico junto a la liberación de RLO. Además, la xantina oxidasa unida a las células endoteliales puede utilizar NADH y mantener junto a la NADPH oxidasa la producción endotelial de O2-. La principal evidencia que implica a la xantina oxidasa en la patogenia de la disfunción endotelial deriva de estudios en los que se ha observado una mejoría en la vasodilatación dependiente del endotelio tras la administración de oxopurinol y alopurinol.
En pacientes con SAHS Sahebjami53 observó que la excreción urinaria de ácido úrico se relacionaba significativamente con el índice de apnea-hipopnea y se normalizaba con el tratamiento con CPAP. Otros estudios también han detectado un aumento de los productos de degradación de las purinas en estos pacientes54-56. Estos resultados indican que la liberación conjunta de RLO que ocurre durante el metabolismo de estas moléculas es probable que contribuya al daño oxidativo relacionado con el SAHS.
Expresión de genes sensibles
La expresión de genes sensibles a un aumento de RLO se acompaña de una activación de factores de transcripción y vías de señalización relacionados con acontecimientos proaterogénicos. Estos factores de transcripción están influidos por el estado redox y la disponibilidad de oxígeno19. El factor 1 inducible por hipoxia regula la transcripción de genes que codifican proteínas que participan en respuestas adaptativas en situaciones de hipoxia, mientras que el factor nuclear *B y la proteína activadora 1 participan en la regulación de genes relacionados con la expresión de citocinas, factores de crecimiento y moléculas de adhesión que están implicados en respuestas inflamatorias y en la progresión de la arteriosclerosis57-59.
Los estudios que indican una activación de estos factores de transcripción en pacientes con SAHS son indirectos y derivados de la detección en plasma de concentraciones elevadas de proteínas codificadas por estos genes, como son el factor de crecimiento vascular endotelial, la endotelina 1, la eritropoyetina, el factor de necrosis tumoral alfa, las interleucinas 1 y 6, la molécula de adhesión intracelular 1, la molécula de adhesión celular vascular 1, la L-selectina y la E-selectina60-69. La producción y liberación de citocinas como el factor de necrosis tumoral alfa y la interleucina 6 podría contribuir a una respuesta inflamatoria sistémica que, a su vez, parece estar relacionada con otro factor de riesgo cardiovascular muy prevalente en estos pacientes como es la obesidad70-73. Por otra parte, también existe evidencia de un aumento de la expresión de moléculas de adhesión (CD15 y CD11) en monocitos aislados de pacientes con SAHS39, lo que refuerza la hipótesis de la importancia funcional de este proceso en la patogenia de las complicaciones cardiovasculares del SAHS y la necesidad de profundizar en el estudio de los mecanismos moleculares implicados en estas alteraciones.
Conclusiones
Diversas líneas de estudio mantienen la hipótesis de que el estrés oxidativo es un importante mecanismo fisipatológico en la enfermedad cardiovascular asociada al SAHS. Sin embargo, actualmente se desconoce cuáles son las principales fuentes celulares que contribuyen a un incremento de RLO en el SAHS. Un mayor conocimiento de estos mecanismos permitirá una intervención terapéutica más efectiva y una prevención del riesgo cardiovascular atribuible al estrés oxidativo en estos pacientes.
Financiado, en parte, por ABEMAR, SEPAR y Fondo de Investigaciones Sanitarias (01/0786, 02/0334) y Red Respira (RTIC C03/11).







