
Pleural involvement in systemic diseases is usually a sign of lesions occurring at other levels. Despite the low incidence (around 1%) of pleural effusions caused by systemic diseases, more often connective tissue diseases, such as rheumatoid arthritis or systemic lupus erythematosus, may present with this. Similarly, vasculitis, such as Wegener's granulomatosis, Churg–Strauss syndrome, or less prevalent diseases, such as adult onset Still's disease, or human adjuvant disease, can also have pleural involvement. Although their incidence is low, it is important to take them into account when making a differential diagnosis of a pleural effusion. In this article, the systemic diseases that include pleural involvement are reviewed, as well as the characteristics of the effusions and their outcome.
La afectación pleural en las enfermedades sistémicas suele ser un reflejo de las lesiones que se producen a otros niveles. A pesar de la baja incidencia de derrame pleural causado por enfermedades sistémicas (alrededor del 1%), las conectivopatías más frecuentes como la artritis reumatoide o el lupus eritematoso sistémico pueden presentarlo. De la misma forma, vasculitis como la granulomatosis de Wegener, el síndrome de Churg–Strauss, enfermedades menos prevalentes como la enfermedad de Still de inicio en el adulto, o la enfermedad adyuvante humana, también pueden tener la pleura afectada. Aunque su incidencia es baja, es importante tenerlas en cuenta a la hora de realizar el diagnóstico diferencial del derrame pleural. En este artículo se revisan las enfermedades sistémicas que cursan con afectación pleural, así como las características del derrame y la evolución del mismo.
Pleural pathologies are often a reflection of underlying lung disease. In localized subpleural inflammatory processes, such as pneumonia or pulmonary infarction, or in diseases with diffuse injury, such as acute respiratory distress syndrome, pleural effusion is usually present due to the extravascular liquid moving along a pressure gradient from the pulmonary interstitium to the pleural space through the mesothelium.1 Therefore, an inflammatory lesion of the pleural or subpleural tissue can produce pleural effusion, whatever its nature may be. In systemic diseases, pleural effusion is caused, presumably, by the increase in capillary permeability resulting from the existing inflammation, be it either of the subpleural tissue or by direct affectation of the pleura. The severity and the persistence of the lesion will determine whether the evolution is towards resolution or towards pleural fibrosis.
The most important feature of the pleural affectation in systemic diseases, especially in connective-tissue diseases (CTD), is the high capillary permeability. The lesion can be caused by direct infiltration of the pleura or by an immune mechanism. It has been shown that in diseases like rheumatoid arthritis (RA) or systemic lupus erythematosus (SLE) there are circulating immune complexes, both in the blood as well as in the pleural liquid (PL), which can be localized in the subpleural tissue or in the pleural capillaries and activate the complement system; this would initiate an endothelial injury that would allow for a liquid rich in proteins to accumulate in the pulmonary interstitium or in the pleural space.2 Additionally, complex reactions are produced, headed by the release of different proteolytic enzymes, from the neutrophils and cytokines from the macrophages, which would not only affect the capillary permeability but might also modulate the migration of the fibroblasts, playing a relevant role in the extension of the pleural lesion.3
The incidence of pleural effusion due to systemic disease is not high when large series are reviewed. In two extensive reviews carried out in Spain, the incidence was approximately 1%.4,5
The aim of this review is to describe the pathogeny, clinical findings and characteristics of the PL of pleural disease associated with systemic diseases.
Rheumatoid ArthritisRA is a chronic inflammatory disease that generally affects the small joints of the feet and hands but can extend to any synovial joint.6 Its diagnostic criteria have been defined since 1987, although these may change as the understanding of the risk factors improves and new molecular markers are developed.7 RA is probably the result of a complex interaction between genetic susceptibility and environmental exposure that provoke an abnormal immune response.
In an extensive review, Cohen and Sahn consider that pleural affectation (pleuritis, effusion, thickening and pneumothorax) is the most frequent intrathoracic manifestation of RA, happening in approximately 5% of patients.8 However, on chest radiography, the pleural involvement is much higher. Juric et al. demonstrate that in 24% of the radiographies of men and 16% of women with RA, there is evidence of pleural disease (thickening and/or pleural effusion).9 Several series have demonstrated that the majority of the patients with RA and pleural effusion correspond with a subtype made up basically of middle-aged men (80%), with high titers of rheumatoid factor (RF), rheumatoid nodules (80%) and a higher prevalence of HLA-B8 and Dw3.9–13
The majority of patients with rheumatoid pleural effusion (RPE) have a small quantity of liquid and are usually asymptomatic.14 The effusions are usually unilateral (in more than 70%), more frequently on the left side, although bilateral and even migratory (from one side to the other) effusions have been reported.15 However, on occasions the pleural effusion increases and can produce thoracic pain (30%–50% of cases), generally with pleuritic characteristics, fever (in one-third of patients, which would require ruling out an additional infection)2 and dyspnea proportional to the quantity of PL. If this does not correspond with the quantity of existing liquid, or if cough appears, it is likely that there is an underlying pulmonary disease.16
RPE usually appears years after the diagnosis of RA and in 25% of cases it can precede or occur simultaneously with the beginning of the joint disease.12 The RPE can be transitory, recurring or chronic,17 in which case it could persist for years.12
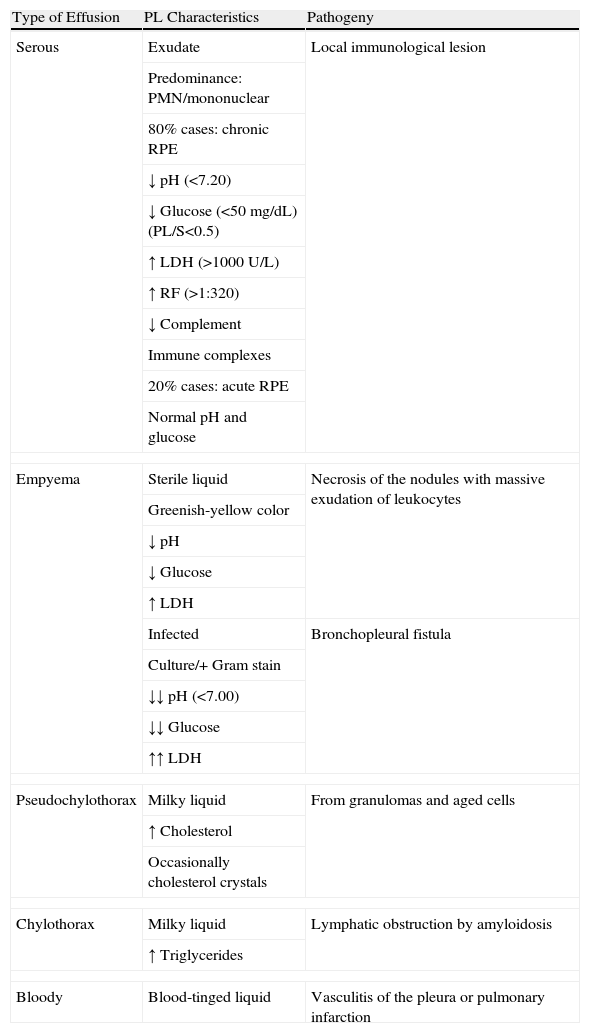
The PL may be serous, cloudy, greenish-yellow, milky or occasionally hemorrhagic in appearance17 (Table 1). Generally, it is an exudate with a high concentration of proteins, and the predominance of nucleated cells will depend on the time elapsed between the initiation of the acute inflammatory process and the moment that thoracocentesis is carried out: there will be a predominance of polymorphonuclear cells when this time is short, and of mononuclear cells in effusions of longer evolution. The typical biochemical characteristics of chronic RPE (80% of cases) are usually pH<7.20, low glucose level (in 80% of cases under 50mg/dL), with a PL/serum ratio <0.5, high levels of lactate dehydrogenase (LDH) (>1000U/L), RF titer higher than 1/320 (generally higher in PL than in blood), total hemolytic complement and low complement components, with an increase in immune complexes in the PL.18 Contrarily, in acute RPE (15%–20% of cases) the pH and glucose levels are usually normal.
Characteristics of Pleural Effusion in Rheumatoid Arthritis.
| Type of Effusion | PL Characteristics | Pathogeny |
| Serous | Exudate | Local immunological lesion |
| Predominance: PMN/mononuclear | ||
| 80% cases: chronic RPE | ||
| ↓ pH (<7.20) | ||
| ↓ Glucose (<50mg/dL) (PL/S<0.5) | ||
| ↑ LDH (>1000U/L) | ||
| ↑ RF (>1:320) | ||
| ↓ Complement | ||
| Immune complexes | ||
| 20% cases: acute RPE | ||
| Normal pH and glucose | ||
| Empyema | Sterile liquid | Necrosis of the nodules with massive exudation of leukocytes |
| Greenish-yellow color | ||
| ↓ pH | ||
| ↓ Glucose | ||
| ↑ LDH | ||
| Infected | Bronchopleural fistula | |
| Culture/+ Gram stain | ||
| ↓↓ pH (<7.00) | ||
| ↓↓ Glucose | ||
| ↑↑ LDH | ||
| Pseudochylothorax | Milky liquid | From granulomas and aged cells |
| ↑ Cholesterol | ||
| Occasionally cholesterol crystals | ||
| Chylothorax | Milky liquid | Lymphatic obstruction by amyloidosis |
| ↑ Triglycerides | ||
| Bloody | Blood-tinged liquid | Vasculitis of the pleura or pulmonary infarction |
LDH: lactate dehydrogenase; PL: pleural liquid; PMN: polymorphonuclear; RPE: rheumatoid pleural effusion; RF: rheumatoid factor; S: serum.
The causes due to which glucose levels in RPE are low are either the block of the entry of glucose to the pleural space through a thickened pleura19 or rather a greater consumption of glucose by an inflamed pleura as it has been observed that fluctuations in the serum levels of glucose do not influence the glucose content in the RPE. The low pH levels reflect active inflammation in the pleural cavity with a high metabolism of glucose and accumulation of lactate and carbon dioxide.
It seems that the reason why pleural effusions are produced in RA is the immunological lesion itself, as it has been demonstrated that while the pleural tissue as well as the mononuclear cells of the PL of patients with RPE synthesize RF, the blood monocytes do not.20
The finding of empyema is not rare in RPE. In the series by Dieppe, 16% of the empyemas were associated with RA.21 Two types of empyemas can be observed: one of them is a sterile liquid with an appearance similar to the empyemas, which is the result of the massive exudation of leukocytes and fibrinoid detritus to the pleural space; another is infected as a consequence of the necrosis of the rheumatoid nodules that, by means of bronchopleural fistula, cause pyopneumothorax.22 In these cases, the pH of the PL can be even lower than usual at around 7.00.23
On occasions, RPE is milky in color (chylous). Chylous RPE (pseudochylothorax) is due to its high lipid content. It is believed that the lysis of hematites and neutrophils in the pleural space releases cholesterol and lecithin–globulin complexes that are trapped in the pleural cavity as a consequence of the pleural fibrotic thickening that blocks the drainage of the liquid that should go out through the parietal lymph nodes.19 However, this theory has recently been put in doubt with the publication of a series of 6 cases with pseudochylothorax secondary to RA with minimal pleural thickening.24 The most frequent causes of pseudochylothorax are long-evolving tuberculous pleural effusions and RPE25 (although there are only 21 reported cases of the latter24) and in both cases the cholesterol values can be higher than 1000mg/dL.
Chylothorax associated with RA has been described. In these cases, the milky pleural exudate presents high levels of triglycerides (>110mg/dL) and are due to lymphatic obstruction due to secondary amyloidosis that on occasion appears in RA.26,27
Bloody pleural effusions are uncommon in RA16 and are secondary to underlying pulmonary infarction due to the existing vasculitis.28
If the RF titer in a pleural exudate is equal or higher than in blood, it is highly suggestive of RPE.2 In these effusions, RA cells or “ragocytes” (leukocytes with phagocytic intracellular inclusions with RF-releasing capability) have also been observed12 but it is not recommended to routinely determine these due to their low specificity. Equally, in the RPE, high levels of SC5b-9 (product of a pathological pathway of activation of the complement system) can be found, along with low levels of C3 and C4 (which entails an autoimmune activation of the inflammatory processes in the RPE),29 high concentrations of ferritin,30 beta-2-microglobulin and angiotensin-converting enzyme,31 neuron specific enolase,32 hyaluronan,33 hydroxyproline34 and adenosine deaminase.35 However, none of these determinations is diagnostic for RPE.36
Although the blood cell count in RPE does not have specific value, the cytology of the pleural liquid can be of great use to support the diagnosis. The characteristic cytology responds to the triad of giant multinucleated macrophages, elongated macrophages and a necrotic background material in the absence of mesothelial cells. However, its specificity has not been evaluated in large series of non-selected PL.37
Generally, the biopsy of the parietal pleura is not usually diagnostic. The characteristic finding is usually the replacement of the normal mesothelial cells for epithelioid cells with giant multinucleated cells12 without there usually being any rheumatoid granulomas observed.38 Pleural biopsy is not usually done routinely and it is indicated in atypical cases of RPE: absence of rhinitis, chylous pleural effusion or suspicion of tuberculosis or malignancy.39
Thoracoscopy can be useful for the evaluation of patients with suspicion of RPE. On the surface of the visceral pleura, non-specific inflammation is observed, while that of the parietal pleura has a granular, slightly inflamed and thickened appearance with numerous small granules that measure around 0.5mm in diameter.12
The evolution of RPE is variable. In the majority of cases, RPE is a small, asymptomatic effusion that does not require any type of intervention unless another alternative diagnosis is suspected. However, its resolution is frequently not complete before 3–4 weeks and usually takes several months. 50% of the cases can be prolonged, which can lead to thickening of the pleura, trapped lung and infection.40 In cases of large symptomatic effusion, treatment can consist of therapeutic thoracocenteses, instillation of intrapleural corticosteroids or fibrinolytics and increased systemic immunosuppression, including oral corticosteroids,10,12,36,41 without any single one of these methods shown to be superior to the rest.36
In refractory effusions, it may be necessary to recur to pleurodesis and even to decortication in cases of pulmonary fibrosis with trapped lung42 in order to alleviate the dyspnea, even though this intervention carries important morbidity and mortality.43 In cases with bronchopleural fistula, it is possible that standard management with pleurodesis by video-assisted thoracoscopic surgery (VATS) is not adequate as the presence of underlying rheumatoid nodules can make local healing difficult. This situation means that the initial closure of the fistula, by thoracotomy, is an option to keep in mind.44
Empyemas are associated with the presence of necrotic subpleural nodules, are usually polymicrobial and should be treated with antibiotics and thoracic drain.36 In these cases, it would be indicated to rule out the presence of a bronchopleural fistula. The preferred drainage method in empyemas with no fibrosis or loculation is the thoracotomy tube, leaving VATS or open thoracotomy as last resort options.45
One uncommon complication of RA is pneumothorax. Its incidence is 6%17 and can be unilateral, bilateral or recurrent. These pneumothoraces take an average of two weeks to resolve, despite chest drainage.46 A group of patients have been reported with the triad of rheumatoid pulmonary disease, pneumothorax and peripheral eosinophilia, with an association between this and disease severity in such a way that a marked eosinophilia could foreshadow a complication like pneumothorax.47
Systemic Lupus ErythematosusSLE is an autoimmune disease that is potentially severe and frequently incapacitating with multiorgan affectation and a fluctuating course with phases of improvement and decline. It is considered as the prototype of autoimmune disease and it is characterized by the production of a wide series of autoantibodies as well as by a variable clinical presentation. Its most frequent initial manifestations are arthritis, photosensitive rash, autoimmune cytopenia and glomerulonephritis.48 It is considered as a disease for women of child-bearing age (ratio 9:1), although the lung involvement is proportionally more frequent in men. In the LUMINA study, when compared with Caucasians, both Afro-Americans and Hispanics had an incidence of SLE that was three times greater, developed earlier and presented greater morbidity and mortality.49
Pleural affectation can be the presentation symptom in 5% of cases,17 although between 30% and 50% of patients with SLE will develop symptomatic pleural inflammation during the course of their disease.50 The typical presentation of pleural affectation in SLE is acute chest pain with pleuritic characteristics that may be accompanied by dyspnea, cough and fever. Unlike RPE, lupus pleuritis is generally symptomatic at the time when diagnosed. Patients with SLE often develop serositis (pleuritis) as part of their disease,51 but effusion can also be due to renal affectation, pulmonary embolism or heart failure.36 Effusions tend to be bilateral but small and may not be evident on chest radiography.52 In contrast with RPE, lupus pleuritis is not accompanied by underlying lung disease.53
In SLE, pleuritis is the result of a process of localized immune inflammation with activation of the complement system and production of immune complexes.54 Pleural effusions behave like typical exudates with high protein levels (>3.5g/dL) and LDH (<500U/L).55 The leukocyte count ranges between 500 and 15,000μL56 and the predominance of nucleated cells can be lymphocytes as well as polymorphonuclear cells. The concentration of glucose is low although not as much as in RPE, and the pH is normally higher than 7.30. The findings with low levels of complement and high titers of antinuclear antibodies (ANA) (>1/160) are suggestive, but not diagnostic, of lupus exudate57,58 as some neoplastic effusions, especially lymphomas, can run their course with high titers.59 On the contrary, the presence of lupus erythematosus (LE) cells is highly specific,60 although this test is rarely carried out due to the long preparation time needed.52 In the pleural biopsy of patients with lupus effusion, a specific pattern of immunofluorescence has been observed that is characterized by the nuclear staining of the pleural cells, either with anti-IgM, anti-IgG or anti-C3.54
There are many medications that can be implicated as a cause of drug-induced lupus,60 and pleural effusion may also be present. The clinical symptomatology in these cases can vary between the mere presence of ANA and florid symptoms of SLE. Once the drug has been withdrawn, the symptoms tend to disappear slowly. In these effusions, even if there is a small renal affectation, the levels of complement are normal,61 although the rest of the cell and biochemical findings are similar to those of SLE.
Although small asymptomatic pleural effusions resolve spontaneously, in the majority of cases the effusions due to SLE or drug-induced lupus respond well to either non-steroid anti-inflammatory preparations or low doses of oral corticosteroids.40 On occasion, it is necessary to manage higher doses of corticosteroids in order to reach the resolution of the effusions. Only very rarely is it necessary to resort to other immunosuppressants in order to control refractory or recurring pleuritis.62 Other treatment methods for this pleuritis include talc pleurodesis63 or tetracycline,64 intravenous immunoglobulin65 or pleurectomy.66
Sjögren's SyndromeSjögren's syndrome (SS) is a chronic inflammatory autoimmune disease that is characterized by a lymphocytic infiltration of the exocrine glands and of multiple extraglandular areas, such as lungs, thyroids, kidneys or hepato-biliary tract.67 The disease can present in an isolated manner (primary SS) or associated with other CTD, more frequently with RA (secondary SS). In less than 5% of the cases, this lymphoproliferation may experience a malign transformation, fundamentally in non-Hodgkin lymphoma. Its pathogeny includes different genetic, environmental and genetic factors. The diagnosis is based on 6 criteria established by a consensus group.68
The pleural involvement includes the presence of effusions (uni- or bilateral), nodules, thickening (associated with recurring pneumonias and atelectasis) and pleural adherences.69 The appearance of pleural effusion is more often associated with SS secondary to RA or SLE. Pleural effusions in primary SS are rare. The PL is an exudate with a high content of B-lymphocytes (fundamentally CD3+ and CD20+), normal levels of pH and glucose and low levels of adenosine deaminase (ADA). The titers of RF and ANA are usually positive, as well as those of anti-SS-A/anti-SS-B antibodies.36 As lymphoma is not infrequent in SS, pleural biopsy should be considered in these patients.70 Effusions usually resolve either spontaneously or with corticosteroids.52
Systemic SclerosisSystemic sclerosis (SS) is an autoimmune disease of the connective tissue of unknown etiology.71 It is infrequent and characterized by presenting organic fibrosis, vasculopathy of the small vessels and specific antibodies of the disease. The organic fibrosis can affect the teguments, gastrointestinal tract, lungs, heart and kidneys.72 There are clear diagnostic criteria73 with various SS subtypes (limited skin SS – previously CREST syndrome; diffuse skin SS – previously progressive systemic scleroderma; and SS without scleroderma).74
SS can affect the pleura by fibrosis or effusion. Effusion is infrequent (7%) but it can reach up to 15% in the scleroderma-overlap syndrome (SS/myositis/RS).75 Pleuritis is more frequent in the diffuse cutaneous SS subgroup and can often be associated with pericardial effusion. Normally the PL is an exudate, but occasionally the effusion is not due to SS itself but instead of chronic renal insufficiency or accompanying heart failure, and therefore could behave as a transudate.75,76
In a postmortem study, pleural thickening and subpleural cystic changes were observed in 86% of the cases.77 Due to the rupture of these subpleural cysts, the appearance of recurring spontaneous pneumothorax is not infrequent78 and which, on occasion, require chest drains over long periods of time.79 Nevertheless, it is surprising that the incidence of pneumothorax is so low, given the subpleural cystic changes that exist. It is speculated that these patients can have pleural symphysis, either partial or complete, as a consequence of previous infectious complications.17
Mixed Connective Tissue DiseaseIn 1972, Sharp et al. described, for the first time, a new clinical entity called mixed connective tissue disease (MCTD) in which the patients presented a combination of clinical features of SS, SLE and inflammatory myopathy, whose diagnosis currently requires three of the following criteria: synovitis or myositis (one of the two), hand edema, Raynaud's phenomenon, acroscleroderma and serologic evidence of positive anti-snRNP in at least one moderate titer.80
It is believed that pleural effusion is produced as a consequence of immunological lesions in the pleura and it tends to be bilateral.81 The overall incidence of pleural effusion in MCTD is 50%. There is some controversy regarding the frequency of pleural effusion in MCTD,82 although in a retrospective study of 81 patients with MCTD, 6% presented pleural effusion and an additional 3% presented pleural thickening.83 The effusions are usually straw-colored exudates with a predominance of polymorphonuclear cells, high levels of proteins (3.5g/dL) and LDH (400U/L) and with completely normal glucose values.84 No complications have been described in the evolution of these effusions.
Ankylosing SpondylitisAnkylosing spondylitis (AS) is a chronic inflammatory disease that predominantly affects the axial skeleton and the peripheral joints in 20% of cases. The affectation of the sacroiliac joints is constant and characteristic. The inflammation of the joints produces pain and progressive rigidity of the spinal column, thorax and pelvis.85 AS can affect the tracheobronchial tree and pulmonary parenchyma and is associated with unique lung manifestations such as the restriction of the chest wall and fibrocystic disease of the upper lobes.86
Pleural affectation is uncommon. In a retrospective study of 2080 patients, pleural effusion was only found in three cases (0.15%), diffuse pleural thickening in one and spontaneous pneumothorax in another,87 although in other series the prevalence almost reaches 5%.88 The pathogeny of pleural effusion is unknown although it has been related with underlying lung disease.89 The effusion is usually a transitory pleural exudate, with normal levels of pH and glucose and a straw-like color. Different predominant nucleated cells have been described,90,91 including eosinophils.92 Pleural thickening is found in almost all patients with fibrocystic diseases and may be due to the chronic inflammatory process or rather to the colonization and micro-invasion of Aspergillus. The radiological finding of progressive pleural thickening should make us contemplate the presence of an aspergilloma.93
As a consequence of apical fibrocystic disease, the probability of spontaneous pneumothorax is greater than in the general population.94 Cases of bilateral pneumothorax have been reported in patients with long-evolution AS.95
Polymyositis/DermatomyositisIdiopathic inflammatory myopathies are chronic acquired autoimmune diseases that cause muscle weakness due to inflammation of the skeletal muscle. They have been classified into three subtypes: polymyositis (PM), dermatomyositis (DM) and inclusion body myositis.96 All have characteristic proximal muscle weakness, high muscle enzymes in serum (especially creatine kinase), electromyographic data of myopathy and infiltrates of inflammatory cells in the muscle tissue. If the patients have manifestations of rash with varying characteristics, it is classified as dermatomyositis.97
The lung is the most frequently affected extramuscular organ in PM and DM.98 Pleural disease in these entities is infrequent99; it does not present in an isolated manner, but instead associated with diffuse interstitial lung disease.100 Due to the fact that DM is often associated with malignancy, it is recommended to always perform thoracocentesis in this type of effusion.101
Pneumothorax and pneumomediastinum can occur in PM and DM as a consequence of the rupture of the alveoli in the framework of a diffuse interstitial lung disease.102
Wegener's GranulomatosisWegener's granulomatosis is a necrotizing granulomatous vasculitis of the small vessels that affects the upper and lower respiratory tracts and the kidneys (glomerulonephritis).103 The lesions in the respiratory tract are present at the onset of the disease in 75%–95% of cases.104
The incidence of pleural effusion varies between 5% and 55% and its pathogeny is not well known. Possible mechanisms involved include subpleural vasculitis that would cause underlying pulmonary parenchyma infarction, heart failure due to hypertension, progressive uremia and bacterial infection of the infarcted lung resulting in parapneumonic effusion. Usually, the effusions are small and unilateral although they can also be bilateral. The liquid is an exudate with a predominance of polymorphonuclear cells without other differential characteristics.105,106 Pleural biopsy can reveal typical granulomatous inflammation with necrosis.107 Pleural effusion is not usually a clinical problem and it resolves either spontaneously or with immunosuppressants.108 The presence of spontaneous pneumothorax and pyopneumothorax, as a result of the rupture of a cavitated nodule, is an uncommon complication.109
Churg–Strauss SyndromeChurg–Strauss syndrome (CSS) is characterized by the clinical triad of asthma, eosinophilia and vasculitis. Lanham et al. describe three phases in CSS: a prodromal phase with asthma, rhinitis and sinusitis; a second phase in which eosinophilia is observed; and a third in which vasculitis appears.110 Two subtypes of CSS have been described, which can overlap: one with positive ANCA and greater renal affectation, and another with negative ANCA that runs its course with a more marked eosinophilia and more frequent and severe cardiac and lung (lung infiltrates) affectation.111
Pleural affectation is frequent. It is estimated that approximately 29% of patients have pleural effusion.17,110 The increase in the permeability of the microcirculation caused by vasculitis and the infarction of the underlying pulmonary parenchyma can play a relevant role in the pathogeny of the pleural effusion in this syndrome. Another theory is that the pleural effusion could be caused by the blockage and dilation of the lymph nodes due to the infiltration of the interlobar connective tissue by the eosinophils.112 The PL is usually bloody with a high percentage of eosinophils and responds well to treatment with oral corticosteroids.113
Behçet's DiseaseBehçet's disease is an inflammatory disease of unknown origin that usually affects young adults and is characterized by recurring oral and genital aphthous ulcers, uveitis and skin lesions.114 The lung affectation is infrequent and ranges between 1% and 7.7%.115,116 Among these patients, pleural effusion in observed in 70% of these cases.
The pathogenic mechanism that produces pleural effusion is due to obstruction secondary to thrombosis of the large central veins (superior vena cava, innominada, subclavian and internal jugular). The pleural effusion may be a transudate (due to obstruction of the superior vena cava that causes an increase in venous pressure),117 or rather chylothorax.118,119 It is argued that chylothorax is produced because the thrombosis of the subclavian vein can obstruct the orifice of the thoracic duct, which leads to an increase in the intraluminal and retrograde pressures of the communicating vessels, with chyle passing through the pleural lymph nodes towards the pleural space.120 The treatment of patients with chylothorax is difficult. While in some cases good response has been obtained with oral corticosteroids,119 in others no response is achieved.118 Therefore, on occasions, it is necessary to resort to drainage with a chest tube followed by chemical pleurodesis121 in addition to anticoagulation and immunosuppressant treatment.
SarcoidosisSarcoidosis is a systemic granulomatous disease that is characterized by a variable presentation and clinical course. Although 90% of the patients have thoracic affectation, any organ may be affected.122 Its diagnosis is based on: (1) compatible clinical and radiological findings; (2) tissue biopsy revealing non-necrotizing epithelioid cell granulomas; and (3) absence of agents capable of producing granulomas.123 Despite the efforts made in recent decades, the etiological agent continues to be unknown.124
Pleural involvement is not frequent in sarcoidosis. Its incidence oscillates between 0.7% and 10%125 and it can present as nodules or pleural thickening, pneumothorax or pleural effusion.126 In a recent study, 5/181 patients (2.8%) diagnosed with sarcoidosis presented pleural effusion, as demonstrated by thoracic ultrasound.125 However, the effusion was only attributed to sarcoidosis in two cases (1.1%). In another series, 25/61 patients with sarcoidosis (41%) had pleural affectation detected by computed chest tomography (CT). Of these, 20 patients presented pleural thickening and 5 effusion.127 Nevertheless, the cause of the pleural effusion was not determined and, in addition, it is probable that the patients were preselected.
The mechanism by which pleural effusion is produced is presumably similar to that of other infiltrative diseases: an increase in capillary permeability due to the pleural affectation. However, other mechanisms may also be involved, such as obstruction of the vena cava, endobronquial sarcoidosis (responsible for bronchial stenosis and lobar atelectasis), trapped lung and lymphatic compression as a cause of chylothorax.126 The pleural effusions are usually small or medium in volume (although massive effusions have also been reported) and are more frequent on the right side (45%) than on the left (33%) or bilateral (22%).126 They may occur in any radiological stage, although the majority of the patients are in stage 2.125
The PL is usually serous, but blood-tinged,128 bloody,129 turbid130 or chylous effusions have also been reported.126 The characteristic analysis of the effusions demonstrates a paucicellular liquid that is predominantly lymphocytic and biochemically corresponds with an exudate and runs its course with levels of glucose similar to those of the serum.125 However, cases of transudates have also been reported,131 with a predominance of eosinophils132 and with low levels of glucose.130 Groman et al. analyzed the lymphocytic subpopulations in the PL and found a high CD4/CD8 ratio, similar to that found in the bronchoalveolar lavage of the patients with active sarcoidosis.133 The definitive diagnosis requires the observation of non-caseating granulomas in the pleural biopsy and the exclusion of other granulomatous diseases of known etiology.
The majority of the effusions are resolved spontaneously in one to three months.8 However, cases have been published of resolution in two weeks after treatment with corticosteroids134 and others in 6 months, with or without the administration of corticosteroids.135 The spontaneous resolution of the effusions correlates well with the absence of symptoms. The treatment with corticosteroids should be kept in mind if the patient is symptomatic or if the effusion is recurring. Cases have also been described of effusions with progression towards chronic pleural thickening or trapped lung that required decortication.128
Eosinophilia–Myalgia SyndromeEosinophilia–myalgia syndrome was described in 1989 when the New Mexico Department of Health (USA) and the Centers for Disease Control (Atlanta, Georgia, USA) published a series of three cases that presented severe myalgia and peripheral eosinophilia after taking medication that contained tryptophan.136,137 The biopsies from the muscle and several abdominal organs revealed eosinophilic infiltration in all of them.
One year later, Swygert et al. described a total of 1531 new cases,138 among which there were 27 deaths. The most frequent symptoms were arthralgia (73%), rash (60%), peripheral edema (59%) and cough or dyspnea (59%). 12% of the 718 patients that underwent chest radiography had pleural effusion, although in a later study this percentage was 33%.139 Generally, the effusions were bilateral and the liquid behaved as a sterile eosinophilic exudate.139 The pleural affectation is not usually clinically significant and the effusion disappears after the standard treatment with high-dose corticosteroids.
Eosinophilic FasciitisEosinophilic fasciitis is an uncommon disease of unknown etiology (although related with intense exercise, start of hemodialysis and infection by Borrelia burgdorferi), described by Shulman in 1974.140 It has a symmetrical affectation and in the initial phase it is characterized by erythema and edema of the limbs and trunk. Later on, it presents an increase in the collagen of the dermis and the subcutaneous fascia. It is usually accompanied by arthralgia, polyclonal hypergammaglobulinemia and eosinophilia. In 10%–20% of cases, the disease remits spontaneously, although it may progress to diffuse cutaneous systemic sclerosis. The response to high doses of prednisolone is good.141
Killen et al. described a case that ran its course with bilateral pleural effusion and presented a great number of inflammatory cells, the majority eosinophils, which disappeared after receiving treatment with prednisolone.142
Angioimmunoblastic T-Cell LymphomaAngioimmunoblastic T-cell lymphoma (AITL) is a lymphoma of peripheral T-cells that is characterized by systemic disease, a polymorphous infiltrate that affects the lymph nodes, with prominent proliferation of high endothelial venules and the follicular dendritic cells. Previously, it was considered an angioimmunoblastic lymphadenopathy with a greater risk of progression towards lymphoma, but the current evidence suggests that AITL is produced as a peripheral T cell lymphoma that represents approximately 15%–20% of all the non-Hodgkin lymphomas. Although a possible role of the Epstein–Barr virus (EBV) in its etiology has been suggested, the neoplastic T-cells are EBV-negative.143
Cullen et al. published a series of 10 cases with angioimmunoblastic lymphadenopathy in which 5 presented ascites and pleural effusion.144 Sugiyama et al. described 5 cases of their own and another 21 collected from the Japanese literature. In their 5 cases, all had pleural effusion, as did 8 of the 21 reviewed patients (50%).145 Neither of the two series provide data about the characteristics of the PL.
Giant Cell ArteritisGiant cell arteritis (GCA) is the most frequent type of systemic vasculitis both in the United States as well as in Europe146 and is characterized by granulomatous affectation of the arteries of large and medium size, with a predilection for the branches of the external carotid artery, especially of the superficial temporal artery.147
GCA does not always debut with the classic manifestations of cephalea, jaw claudication and blindness. In 9% of patients, the respiratory symptoms are present in the beginning of the disease and in 4% they are the first manifestation.148 Pleural effusion is a very uncommon form of presentation and only isolated cases have been published.149–151 The PL is usually an exudate with a predominance of polymorphonuclear cells and with glucose levels similar to those of serum. However, the case described by Ramos et al.150 has a predominance of lymphocytes (70%) and that of Gur et al. is a transudate.152 The histological study of the pleura in patients with effusions associated with temporal arteritis is limited to the samples obtained by closed pleural biopsy. The changes observed in these samples are non-specific, although intense mesothelial reactions have been seen. Due to the size of the samples obtained, the presence of vascular changes cannot be excluded. The response to treatment with corticosteroids is good and the effusion usually disappears.
Kawasaki DiseaseKawasaki disease is a self-limiting, acute systemic vasculitis of unknown origin that affects children. The incidence in Japan is 175 cases/100000 children under the age of 5, while in the United States it ranges between 20 and 25/100000. Its most important complication is the development of lesions in the coronary arteries that vary between transitory dilation and the destruction of the architecture of the vessel wall with the production of aneurisms.153
In a series of 129 patients, Umezawa et al. found that the chest radiograph was normal in 19 (14.7%). Of these, 3 (15.8%) had pleural effusion.154 Although the authors did not analyze the PL, they ruled out that the effusion was due to heart failure as the ultrasounds showed good function of the left ventricle in all the patients. In their opinion, the alterations of the chest radiograph, including the pleural effusion, could be due to the inflammation of the lower respiratory tract, or rather the pulmonary arteritis that is observed in 45%–71% of the cases that undergo autopsy.155
Human Adjuvant DiseaseFrom 1964, cases have been described of connective tissue diseases that develop in patients who have previously undergone plastic surgery with paraffin or silicone implants. This has been called human adjuvant disease due to the fact that the substance injected acts as an adjuvant in the pathogenesis of the disease.156 Although cases have been observed of SLE, RA, MCTD and Hashimoto's thyroiditis, the majority have scleroderma.
Walsh et al. reported a case in which, after mammoplasty with silicone prostheses, SLE was induced which had associated bilateral chylous effusion.157 It is known that 15% of the chylous effusions are idiopathic26 and this could be a case, but the authors are inclined to think that this effusion was due to the SLE because it was accompanied by pericarditis and a high ANA titer. Clinical improvement presented with the disappearance of the effusion as well as negative ANA titer after the withdrawal of the prostheses.
Adult-Onset Still's DiseaseAdult-onset Still's disease (AOSD) is an uncommon systemic inflammatory disease of unknown etiology that affects young adults of both sexes between the ages of 16 and 35. Its diagnosis is difficult as there is no single diagnostic test (although serum ferritin is usually very high) or specific histopathological characteristics,158 which has led to the development of different classifications for the diagnosis of the disease. In this direction, several authors consider the presence of pleuritis or pericarditis as a minor diagnostic criterion.159
Pleuritis is the most frequent pulmonary manifestation. It can be observed in the initial presentation, but it is more common for it to become manifest during an exacerbation. The pleural effusions are usually bilateral and the PL behaves like an exudate with a predominance of neutrophils.160 It sometimes forms part of a generalized serositis (pericardial effusion and ascites); therefore, given this situation, the differential diagnosis should always include this entity.161
Polyarteritis NodosaPolyarteritis nodosa (PAN) is a systemic necrotizing vasculitis that predominantly affects medium-sized arteries and, although in the majority of patients it is primary in origin, it can be secondary to viral infections, fundamentally by the hepatitis B virus (HBV).162
Pagnoux et al. studied a series of 348 patients with PAN, 12 (3.4%) of whom presented pleural effusion. This was manifested both in the PAN related with HBV (5 cases) as well as unrelated PAN (7 cases).163 This study, designed to describe the main characteristics of the long-term results of the patients with a well-established diagnosis of PAN, does not describe either the size or the location of the effusions, nor does it describe either the characteristics of the PL or its evolution once treatment is initiated.
Polyangiitis Overlap SyndromesThe term “polyangiitis overlap syndrome” was proposed by Leavitt and Fauci to define a type of systemic vasculitis that either could not be included in a single category of the classification of vasculitis, or which overlapped several of them.164 It is characterized by presenting typical manifestations of systemic vasculitis with affectation of multiple organs, eosinophilia and good response to treatment with corticosteroids.
Koarada et al. reported a patient with this disease who presented left pleural effusion that was cloudy and yellowish in appearance. Biochemically, it was an exudate, with low ADA and a predominance of eosinophils (72%). The effusion disappeared after treatment with corticosteroids.165
Conflict of InterestThe authors declare having no conflict of interests.
We would like to thank Dr. Juan Díaz Garel for reviewing the manuscript.
Please cite this article as: Ferreiro L, et al. Enfermedades sistémicas y pleura. Arch Bronconeumol. 2011;47:361–70.







