














La hipertensión pulmonar es un trastorno hemodinámico definido por el aumento anómalo de la presión arterial pulmonar, que puede presentarse en numerosas enfermedades y situaciones clínicas. Las causas de hipertensión pulmonar se clasifican en 5 grandes grupos: arterial, debida a cardiopatía izquierda, debida a enfermedad pulmonar y/o hipoxemia, tromboembólica crónica y de mecanismo no establecido y/o multifactorial. El presente documento expone de forma resumida las recomendaciones de la Guía de Diagnóstico y Tratamiento de la Hipertensión Pulmonar de la Sociedad Española de Neumología y Cirugía Torácica. En dicha guía se presentan las pautas actuales de diagnóstico y tratamiento de los distintos grupos de hipertensión pulmonar.
Pulmonary hypertension is a hemodynamic disorder defined by abnormally high pulmonary artery pressure that can occur in numerous diseases and clinical situations. The causes of pulmonary hypertension are classified into 5 major groups: arterial, due to left heart disease, due to lung disease and/or hypoxemia, chronic thromboembolic, with unclear and/or multifactorial mechanisms. This is a brief summary of the Guidelines on the Diagnostic and Treatment of Pulmonary Hypertension of the Spanish Society of Pulmonology and Thoracic Surgery. These guidelines describe the current recommendations for the diagnosis and treatment of the different pulmonary hypertension groups.
En el presente documento se exponen resumidas las recomendaciones de la Guía de Diagnóstico y Tratamiento de la Hipertensión Pulmonar elaborada por la Sociedad Española de Neumología y Cirugía Torácica1, que fue confeccionada a partir de la guía de práctica clínica de la European Society of Cardiology y la European Respiratory Society2. Para mayor detalle remitimos al lector al documento original1 (https://issuu.com/separ/docs/normativa_70?e=3049452/44188557). Los niveles de evidencia y la clase de recomendación empleados se muestran en la tabla 1.
Niveles de evidencia y clases de recomendación empleadas en la Normativa
| Niveles de evidencia | |
| A | Datos derivados de múltiples ensayos clínicos aleatorizados o metaanálisis |
| B | Datos derivados de un único ensayo clínico aleatorizado o estudios no aleatorizados de gran tamaño |
| C | Consenso de la opinión de expertos y/o estudios de pequeño tamaño, retrospectivos o registros |
| Clases de recomendación | |
| I | Evidencia y/o acuerdo general de que un determinado tratamiento o procedimiento es beneficioso, útil o efectivo |
| II | Evidencia conflictiva y/o divergencia de opiniones acerca de la utilidad/eficacia de un determinado tratamiento o procedimiento |
| IIa | La evidencia/opinión se inclina a favor de la utilidad/eficacia |
| IIb | La utilidad/eficacia está menos establecida por evidencia/opinión |
| III | Evidencia o acuerdo general de que un determinado tratamiento o procedimiento no es útil/efectivo y, en algunos casos, puede ser perjudicial |
La hipertensión pulmonar (HP) es un trastorno fisiopatológico y hemodinámico definido por el aumento de la presión arterial pulmonar media (PAPm)≥25mmHg, medida por cateterismo cardiaco derecho (CCD)2. La HP puede presentarse en distintos procesos clínicos, que se clasifican en 5 grupos (tabla 2).
Clasificación de la hipertensión pulmonar (European Society of Cardiology/European Respiratory Society, 2015)
| 1. Hipertensión arterial pulmonar (HAP) |
| 1.1. Idiopática |
| 1.2. Hereditaria |
| 1.2.1. Mutación BMPR2 |
| 1.2.2. Otras mutaciones |
| 1.3. Inducida por fármacos y tóxicos |
| 1.4. Asociada a: |
| 1.4.1. Enfermedad del tejido conectivo |
| 1.4.2. Infección por VIH |
| 1.4.3. Hipertensión portal |
| 1.4.4. Cardiopatía congénita |
| 1.4.5. Esquistosomiasis |
| 1’. Enfermedad venooclusiva pulmonar y/o hemangiomatosis capilar pulmonar |
| 1’.1. Idiopática |
| 1’.2. Hereditaria |
| 1’.2.1. Mutación EIF2AK4 |
| 1’.2.2. Otras mutaciones |
| 1’.3. Inducida por fármacos, toxinas y radiaciones |
| 1’.4. Asociada a: |
| 1’.4.1. Enfermedad del tejido conectivo |
| 1’.4.2. Infección por VIH |
| 1”. Hipertensión pulmonar persistente del recién nacido |
| 2. Hipertensión pulmonar debida a cardiopatía izquierda |
| 2.1. Disfunción sistólica del ventrículo izquierdo |
| 2.2. Disfunción diastólica del ventrículo izquierdo |
| 2.3. Enfermedad valvular |
| 2.4. Obstrucción congénita/adquirida del tracto de entrada/salida del ventrículo izquierdo y miocardiopatías congénitas |
| 2.5. Estenosis congénita o adquirida de las venas pulmonares |
| 3. Hipertensión pulmonar debida a enfermedad pulmonar y/o hipoxemia |
| 3.1. Enfermedad pulmonar obstructiva crónica |
| 3.2. Enfermedad pulmonar intersticial difusa |
| 3.3. Otras enfermedades pulmonares con patrón mixto restrictivo y obstructivo |
| 3.4. Trastornos respiratorios del sueño |
| 3.5. Hipoventilación alveolar |
| 3.6. Exposición crónica a grandes alturas |
| 3.7. Anomalías del desarrollo pulmonar |
| 4. Hipertensión pulmonar tromboembólica crónica y otras obstrucciones de las arterias pulmonares |
| 4.1. Hipertensión pulmonar tromboembólica crónica |
| 4.2. Otras obstrucciones de las arterias pulmonares: |
| 4.2.1. Angiosarcoma |
| 4.2.2. Otros tumores intravasculares |
| 4.2.3. Arteritis |
| 4.2.4. Estenosis congénitas de las arterias pulmonares |
| 4.2.5. Parasitosis (hidatidosis) |
| 5. Hipertensión pulmonar de mecanismo no establecido y/o multifactorial |
| 5.1. Enfermedades hematológicas: anemia hemolítica, trastornos mieloproliferativos, esplenectomía |
| 5.2. Enfermedades sistémicas: sarcoidosis, histiocitosis pulmonar, linfangioleiomiomatosis, neurofibromatosis |
| 5.3. Trastornos metabólicos: enfermedad del almacenamiento del glucógeno, enfermedad de Gaucher, trastornos tiroideos |
| 5.4. Otros: microangiopatía pulmonar tumoral trombótica, mediastinitis fibrosante, insuficiencia renal crónica (con/sin diálisis), hipertensión pulmonar segmentaria |
La ecocardiografía transtorácica (ETT) constituye la herramienta fundamental para la detección y el cribado de la HP. La probabilidad de presentar HP en función de los hallazgos en la ETT se muestra en la tabla 3.
Probabilidad de presentar hipertensión pulmonar a partir de los resultados del estudio con ecocardiografía transtorácica
| Baja | VRT≤2,8m/s o no medible |
| Intermedia | VRT 2,9-3,4m/s; o VRT≤2,8m/s o no medible, en presencia de otros signos ecográficos de hipertensión pulmonar |
| Alta | VRT>3,4m/s; o VRT 2,9-3,4m/s, en presencia de otros signos ecográficos de hipertensión pulmonar |
| * Otros signos ecocardiográficos que indican hipertensión pulmonar: |
| Ventrículos |
| Relación del diámetro VD/VI basal>1,0 |
| Aplanamiento del septo interventricular (índice de excentricidad del VI>1,1 en sístole o diástole) |
| Arteria pulmonar |
| Tiempo de aceleración doppler del tracto de salida del VD<105ms y/o muesca mesosistólica |
| Velocidad de regurgitación pulmonar en protodiástole>2,2m/s |
| Diámetro de la AP>25mm |
| Vena cava inferior y aurícula derecha |
| Diámetro de la vena cava inferior>21mm con disminución del colapso inspiratorio (<50% en inspiración profunda o<20% en inspiración normal) |
| Área de la aurícula derecha (telesistólica)>18cm2 |
AP: arteria pulmonar; VD: ventrículo derecho; VI: ventrículo izquierdo; VRT: velocidad de regurgitación tricuspídea.
Se recomienda el cribado de HP con ETT en sujetos asintomáticos pertenecientes a los siguientes grupos de riesgo:
- –
Pacientes con esclerosis sistémica [I, B].
- –
Familiares de primer grado de pacientes diagnosticados de hipertensión arterial pulmonar (HAP) hereditaria [I, C].
- –
Pacientes con hipertensión portal candidatos a trasplante hepático [I, B].
En el resto de los casos, la ETT se realizará con base en la sospecha clínica.
Aproximación general al diagnósticoLa figura 1 muestra el algoritmo diagnóstico de la HP. Si existe sospecha de HP se realizará una ETT. Si la probabilidad de HP es intermedia o alta se descartará enfermedad cardiaca izquierda (HP grupo 2) y enfermedad respiratoria crónica (HP grupo 3). Los pacientes de dichos grupos con HP o disfunción del ventrículo derecho severas se derivarán a una unidad experta en HP2 [IIa, C]. Excluida la HP de los grupos 2 y 3, la gammagrafía pulmonar de ventilación-perfusión separar palabras se empleará para descartar enfermedad tromboembólica. Si existen defectos de perfusión en la gammagrafía pulmonar de ventilación-perfusión se completará el estudio de probable hipertensión pulmonar tromboembólica crónica (HPTEC). El diagnóstico hemodinámico con CCD se realizará en una unidad experta en HP [I, B]. Si se confirma HAP, se completará el estudio para la identificación del subtipo.

Algoritmo diagnóstico de la hipertensión pulmonar.
CCD: cateterismo cardiaco derecho; ECC: enfermedad cardiaca congénita; ECG: electrocardiograma; ETC: enfermedades del tejido conectivo; ETT: ecocardiografía transtorácica; EVOP: enfermedad venooclusiva pulmonar; HAP: hipertensión arterial pulmonar; HCP: hemangiomatosis capilar pulmonar; HP: hipertensión pulmonar; HPTEC: hipertensión pulmonar tromboembólica crónica; PAPm: presión arterial pulmonar media; PAWP: presión de oclusión arterial pulmonar; PFR: pruebas funcionales respiratorias; RVP: resistencia vascular pulmonar; Rx: radiografía; TC: tomografía computarizada; UW: unidades Wood; V-Q: ventilación-perfusión; VD: ventrículo derecho; VIH: virus de la inmunodeficiencia humana.
En los pacientes con HAP idiopática, hereditaria o asociada a fármacos se efectuará una prueba vasodilatadora con óxido nítrico inhalado o epoprostenol iv durante el CCD diagnóstico [I, C]. Dicha prueba es positiva cuando la PAPm desciende≥10mmHg hasta alcanzar un valor≤40mmHg, sin que disminuya el gasto cardiaco [I, C]. La identificación del subtipo se efectuará mediante ecocardiografía de contraste, análisis de autoinmunidad, serología de virus hepatotropos y serología VIH (fig. 1). En el caso de historia familiar de HP, o cuando se sospeche esta posibilidad, se aconseja efectuar estudio de mutaciones del gen BMPR23–5.
El diagnóstico de enfermedad venooclusiva pulmonar (EVOP) o de hemangiomatosis capilar pulmonar (HCP) se basará en datos clínicos, capacidad de difusión de CO muy reducida, hipoxemia grave y hallazgos compatibles en la TC de alta resolución6. También puede diagnosticarse por la presencia de mutaciones del gen EIF2AK46.
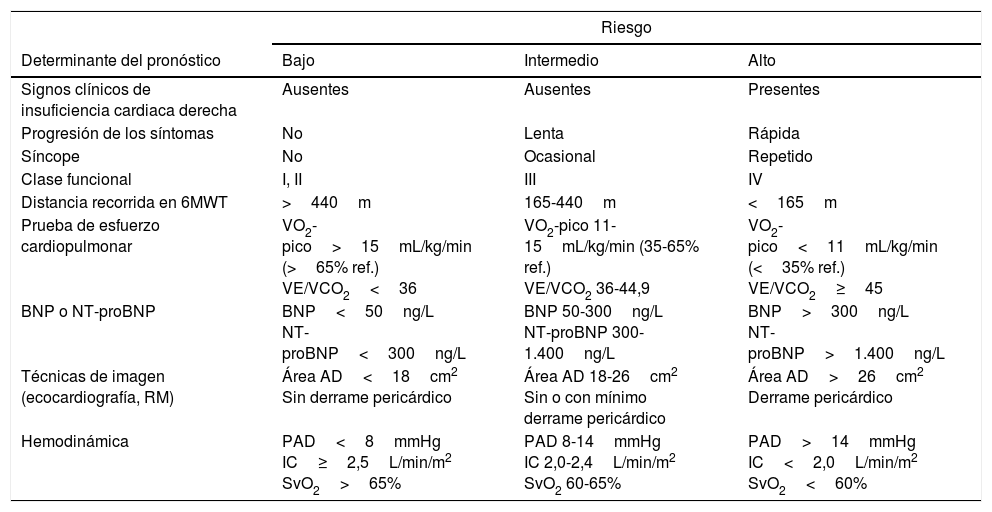
Para la evaluación pronóstica se emplea un conjunto de variables relacionadas con la supervivencia2 (tabla 4). Para el seguimiento, se recomienda que los parámetros clínicos y los de realización más sencilla (clase funcional [CF], prueba de marcha de 6 minutos, ECG, analítica) se evalúen cada 3-6 meses, mientras que los de mayor complejidad se valorarán cada 6-12 meses7, o cuando exista deterioro clínico [I, C].
Evaluación pronóstica en la hipertensión arterial pulmonar
| Riesgo | |||
|---|---|---|---|
| Determinante del pronóstico | Bajo | Intermedio | Alto |
| Signos clínicos de insuficiencia cardiaca derecha | Ausentes | Ausentes | Presentes |
| Progresión de los síntomas | No | Lenta | Rápida |
| Síncope | No | Ocasional | Repetido |
| Clase funcional | I, II | III | IV |
| Distancia recorrida en 6MWT | >440m | 165-440m | <165m |
| Prueba de esfuerzo cardiopulmonar | VO2-pico>15mL/kg/min (>65% ref.) VE/VCO2<36 | VO2-pico 11-15mL/kg/min (35-65% ref.) VE/VCO2 36-44,9 | VO2-pico<11mL/kg/min (<35% ref.) VE/VCO2≥45 |
| BNP o NT-proBNP | BNP<50ng/L NT-proBNP<300ng/L | BNP 50-300ng/L NT-proBNP 300-1.400ng/L | BNP>300ng/L NT-proBNP>1.400ng/L |
| Técnicas de imagen (ecocardiografía, RM) | Área AD<18cm2 Sin derrame pericárdico | Área AD 18-26cm2 Sin o con mínimo derrame pericárdico | Área AD>26cm2 Derrame pericárdico |
| Hemodinámica | PAD<8mmHg IC≥2,5L/min/m2 SvO2>65% | PAD 8-14mmHg IC 2,0-2,4L/min/m2 SvO2 60-65% | PAD>14mmHg IC<2,0L/min/m2 SvO2<60% |
AD: aurícula derecha; BNP: péptido natriurético cerebral; IC: índice cardiaco; NT-proBNP: prohormona N-terminal del péptido natriurético cerebral; PAD: presión de la aurícula derecha; RM: resonancia magnética; SvO2: saturación de oxígeno en sangre venosa mezclada; VE/VCO2: relación entre ventilación minuto y producción de CO2; VO2-pico: consumo pico de oxígeno; 6MWT: prueba de marcha de 6 minutos.
Las medidas generales de tratamiento de la HAP se muestran en la tabla 5. Los diuréticos están indicados en los pacientes con fracaso ventricular derecho y retención hídrica [I, C]. Se emplean diuréticos de asa o antagonistas de la aldosterona2. Se recomienda la anticoagulación con antagonistas de la vitamina K en la HAP idiopática, hereditaria o debida a anorexígenos [IIb, C]. Es aconsejable la oxigenoterapia si la PaO2 es<60mmHg [I, C]. También puede considerarse para la corrección de la desaturación durante el ejercicio2. Se recomienda seguimiento regular de los niveles de hierro y administrar suplementos en caso necesario.
Medidas generales de tratamiento en la hipertensión arterial pulmonar
| Medidas recomendadas [clase I] |
| Evitar el embarazo |
| Prevención de las infecciones |
| Soporte psicosocial |
| Medidas que se deben tener en consideración [clase IIa] |
| Entrenamiento supervisado [evidencia B] |
| Oxigenoterapia en los viajes de larga duración en avión |
| La cirugía electiva mayor debería ser realizada en centros de referencia con experiencia en hipertensión pulmonar |
| Medidas que se pueden tener en consideración [clase IIb] |
| Consejo genético en unidades especializadas en los casos o familiares con mutaciones asociadas a la HAP o la EVOP |
| Evitar los fármacos que pueden agravar la HP (descongestionantes nasales y betabloqueantes) |
| Dieta: aconsejar una ingesta de sal diaria<5g (equivalente a 2g de sodio), en particular en los pacientes con insuficiencia cardiaca derecha. Si esta es severa o existe hiponatremia es aconsejable reducir también la ingesta hídrica a<1,5-2L/día |
| Medidas desaconsejadas [clase III] |
| Actividad física extenuante |
| Subir a altitudes superiores a los 1.500-2.000m sin oxígeno suplementario |
EVOP: enfermedad venooclusiva pulmonar; HAP: hipertensión arterial pulmonar.
Todas las recomendaciones tienen un grado de evidencia C, salvo si se indica lo contrario.
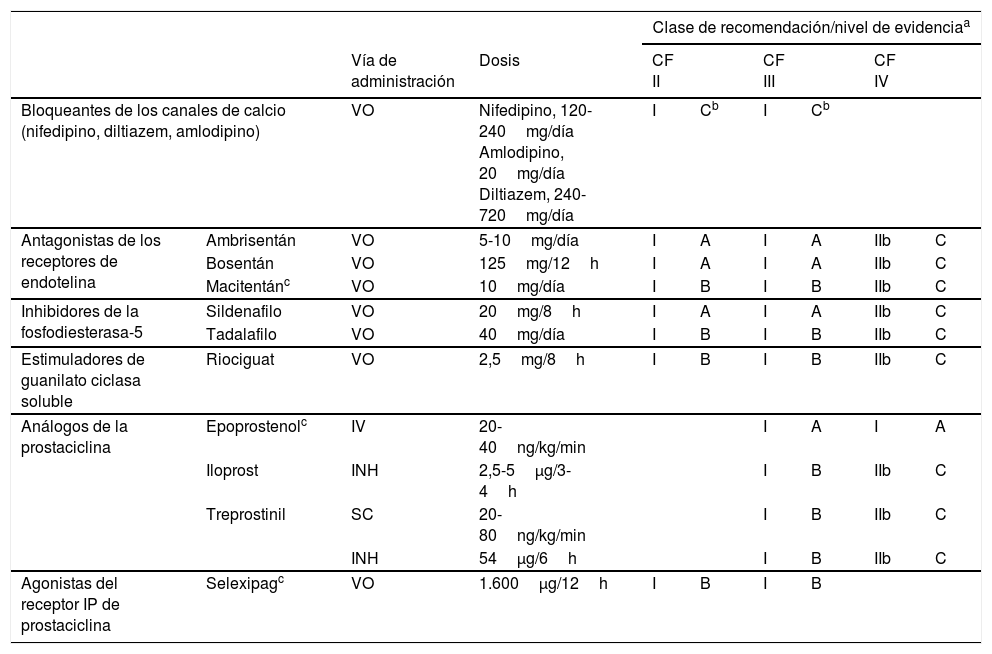
Los fármacos específicos para el tratamiento de la HAP incluyen (tabla 6):
- –
Bloqueantes de los canales del calcio: indicados en pacientes con HAP idiopática y prueba vasodilatadora positiva [I, C]. Se recomienda emplear nifedipino, diltiazem o amlodipino a dosis elevadas8.
- –
Antagonistas de los receptores de la endotelina: incluyen ambrisentán, bosentán y macitentán. Los 2 primeros pueden producir toxicidad hepática, por lo que es obligatorio el control mensual de enzimas hepáticas. Con macitentán se recomienda medir periódicamente el nivel de hemoglobina por el riesgo de anemia.
- –
Inhibidores de la fosfodiesterasa tipo 5 (IPDE5) y estimuladores de la guanilato ciclasa soluble (EGCs): los IPDE5 disponibles son sildenafilo y tadalafilo, y el único EGCs disponible es riociguat. Está contraindicada la administración conjunta de IPDE5 y EGCs.
- –
Análogos de la prostaciclina y agonistas de los receptores de la prostaciclina: dentro de los análogos de la prostaciclina se dispone de epoprostenol, que se administra por vía iv en infusión continua; iloprost, que se administra por vía inhalada; y treprostinil, que se administra por vía sc mediante una bomba de microinfusión continua. También se han observado efectos favorables con treprostinil inhalado9 (tabla 4). Selexipag es un agonista de los receptores de la prostaciclina que se administra por vo10.
Recomendaciones para el tratamiento con monoterapia en la hipertensión arterial pulmonar
| Clase de recomendación/nivel de evidenciaa | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Vía de administración | Dosis | CF II | CF III | CF IV | |||||
| Bloqueantes de los canales de calcio (nifedipino, diltiazem, amlodipino) | VO | Nifedipino, 120-240mg/día Amlodipino, 20mg/día Diltiazem, 240-720mg/día | I | Cb | I | Cb | |||
| Antagonistas de los receptores de endotelina | Ambrisentán | VO | 5-10mg/día | I | A | I | A | IIb | C |
| Bosentán | VO | 125mg/12h | I | A | I | A | IIb | C | |
| Macitentánc | VO | 10mg/día | I | B | I | B | IIb | C | |
| Inhibidores de la fosfodiesterasa-5 | Sildenafilo | VO | 20mg/8h | I | A | I | A | IIb | C |
| Tadalafilo | VO | 40mg/día | I | B | I | B | IIb | C | |
| Estimuladores de guanilato ciclasa soluble | Riociguat | VO | 2,5mg/8h | I | B | I | B | IIb | C |
| Análogos de la prostaciclina | Epoprostenolc | IV | 20-40ng/kg/min | I | A | I | A | ||
| Iloprost | INH | 2,5-5μg/3-4h | I | B | IIb | C | |||
| Treprostinil | SC | 20-80ng/kg/min | I | B | IIb | C | |||
| INH | 54μg/6h | I | B | IIb | C | ||||
| Agonistas del receptor IP de prostaciclina | Selexipagc | VO | 1.600μg/12h | I | B | I | B | ||
INH: inhalada; IV: intravenosa; SC: subcutánea; VO: vía oral.
- –
Septostomía auricular: indicada en pacientes en CF IV, con fallo del ventrículo derecho y síncope11, o como tratamiento puente en espera de trasplante [IIb, C]. Se realizará en centros con experiencia. Debe evitarse en pacientes con presión de la aurícula derecha>20mmHg o con SaO2<85% respirando aire ambiente.
- –
Trasplante pulmonar: habitualmente se realiza trasplante bipulmonar. Está indicado en pacientes jóvenes sin comorbilidad asociada, cuando no existe respuesta al máximo tratamiento médico2 [I, C]. Es el tratamiento de elección en la EVOP y la HCP.
La estrategia de tratamiento de la HAP tiene 4 componentes (fig. 2):

Establecer objetivos terapéuticos: el objetivo principal es que el paciente se encuentre en una situación de bajo riesgo de mortalidad (tabla 4) [I, C]. El perfil de bajo riesgo se caracteriza por buenas tolerancia al esfuerzo, calidad de vida y función ventricular derecha. El riesgo se definirá al inicio, antes de empezar el tratamiento, y en los seguimientos periódicos [I, C].
Aproximación inicial: incluye las medidas generales (tabla 5) y el tratamiento de soporte. El diagnóstico hemodinámico con prueba vasodilatadora debe efectuarse en una unidad experta en HP [I, B], ya que el resultado contribuirá a definir el perfil de riesgo y establecer la pauta de tratamiento.
Tratamiento inicial: en los pacientes con respuesta vasodilatadora positiva se iniciará tratamiento con bloqueantes de los canales de calcio a dosis elevadas. Si a los 3 meses la respuesta clínica es inadecuada se emplearán otros fármacos específicos.
En los pacientes con riesgo bajo o intermedio con respuesta vasodilatadora negativa se iniciará tratamiento con fármacos específicos en monoterapia o combinados (fig. 2). Los antagonistas de los receptores de la endotelina, los IPDE5 y los EGCs en monoterapia han demostrado eficacia en pacientes en CF II y III. Los fármacos prostanoides solo han sido evaluados en pacientes en CF III. La elección del fármaco se basará en la vía de administración, el perfil de seguridad, la posible interacción con otros fármacos, las comorbilidades, la cantidad y la calidad de las evidencias disponibles, las preferencias del paciente, la experiencia del médico y el coste.
Si se opta por el tratamiento combinado de inicio, la única combinación que ha demostrado ser superior al tratamiento con monoterapia es la de ambrisentán y tadalafilo12 [I, B].
En los pacientes con perfil de riesgo alto o en CF IV, el tratamiento de elección es epoprostenol iv13 [I, A]. Existen evidencias de que el tratamiento combinado de inicio de epoprostenol junto con uno o 2 fármacos es eficaz14 [IIa, C].
Valoración de la respuesta: se evaluará la respuesta al tratamiento a los 3-4 meses [I, C]. Si no es satisfactoria, se añadirá un segundo o tercer fármaco y se considerará referir al paciente para evaluación de trasplante pulmonar2. Todos los pacientes deben ser seguidos periódicamente en una unidad experta en HP. La periodicidad de las visitas se establecerá de acuerdo con la gravedad de la enfermedad; en cualquier caso, no debería ser superior a los 6 meses, incluso en los pacientes con respuesta clínica satisfactoria2.
Consideraciones según el subtipoCardiopatías congénitasLas cardiopatías congénitas se incluyen en los grupos 1, 2, 3 y 5 de HP, dependiendo del mecanismo subyacente. La tabla 7 muestra la clasificación de la HAP asociada a cardiopatía congénita y la tabla 8 resume las recomendaciones de tratamiento farmacológico. Se proponen los siguientes límites para el cierre del cortocircuito sistémico-pulmonar [IIa, C]: si el índice de resistencia vascular pulmonar es<4 unidades Wood·m2 está indicado; si es>8 unidades Wood·m2 está contraindicado. En las situaciones intermedias se valorará individualmente15.
Clasificación clínica de la hipertensión arterial pulmonar asociada a cardiopatía congénita
| 1. Síndrome de Eisenmenger | Incluye los defectos intra- y extracardiacos grandes, los cuales empiezan con cortocircuito sistémico-pulmonar y con el tiempo progresan a una elevación de la RVP grave e inversión del cortocircuito (pulmonar-sistémico) o cortocircuito bidireccional. La cianosis, el compromiso multiorgánico y la poliglobulia acostumbran a estar presentes |
| 2. HAP asociada a cortocircuitos sistémico-pulmonares prevalentes | Corregiblesa No corregibles Se incluyen defectos moderados o grandes. La RVP está leve o moderadamente elevada y prevalece el cortocircuito sistémico-pulmonar. La cianosis en reposo no es característica |
| 3. HAP con defectos pequeños o casualesb | Elevación marcada de la RVP en presencia de pequeños defectos cardiacos (habitualmente defectos del septo interventricular<1cm o del septo interauricular<2cm de diámetro, evaluados por ecocardiograma). El cuadro clínico es muy similar al de la HAP idiopática. El cierre de los defectos está contraindicado |
| 4. HAP asociada a CC con defecto cardiaco corregido | La CC se repara pero la HAP persiste inmediatamente después de la corrección o recurre o se desarrolla meses o años después de la misma |
CC: cardiopatía congénita; HAP: hipertensión arterial pulmonar; RVP: resistencia vascular pulmonar.
El tamaño hace referencia a pacientes adultos. Sin embargo, también en los adultos el diámetro puede no ser suficiente para definir la relevancia hemodinámica del defecto o del gradiente de presión, la direccionalidad o el tamaño del cortocircuito, por lo que se debe tener en cuenta la relación entre los flujos pulmonar y sistémico.
Tratamiento farmacológico de la hipertensión arterial pulmonar asociada a cardiopatía congénita
| 1. La anticoagulación está restringida a los pacientes con arritmias auriculares y/o trombosis de las arterias pulmonares [IIb, C] |
| 2. El oxígeno suplementario está indicado si implica mejoría clínica y de la SaO2 [IIa, C] |
| 3. El suplemento de hierro debe considerarse en presencia de ferropenia [IIb, C] |
| 4. Bosentán es el tratamiento de elección en los pacientes con síndrome de Eisenmenger [I, B] |
| 5. El tratamiento combinado con ARE, IPDE5 y/o prostanoides está indicado [IIa, C] |
| 6. El trasplante pulmonar con el cierre del defecto está indicado en las cardiopatías congénitas simples y el cardiopulmonar en las complejas |
ARE: antagonista de los receptores de endotelina; IPDE5: inhibidores de la fosfodiesterasa tipo 5; SaO2: saturación de oxígeno en sangre arterial.
[Clase de recomendación, nivel de evidencia].
La HAP asociada a esclerosis sistémica es la forma de presentación más frecuente16. Se recomienda realizar cribado anual con ETT y capacidad de difusión de CO en los pacientes con esclerosis sitémica17 [I, C]. En el resto de las enfermedades del tejido conectivo, la ETT se recomienda en presencia de síntomas. La TCAR de tórax permite evaluar la presencia de enfermedad pulmonar intersticial y/o EVOP18. El CCD se recomienda en todos los casos de sospecha de HAP [I, C]. Los pacientes con esclerodermia y PAPm entre 21-24mmHg deben ser monitorizados por su alto riesgo de desarrollar HAP19.
El tratamiento de los pacientes con enfermedades del tejido conectivo y HAP debe seguir el algoritmo general de la HAP [I, C]. La anticoagulación oral condiciona un peor pronóstico20, por lo que solo se empleará en casos con predisposición a la trombofilia (anticuerpos antifosfolipídicos)21 [IIb, C]. El tratamiento inmunosupresor puede beneficiar a los pacientes con HAP asociada a lupus eritematoso sistémico o enfermedad mixta del tejido conectivo22.
Hipertensión portopulmonarViene definida por la asociación de hipertensión portal e HP. Los pacientes con hipertensión portopulmonar tienen mayor mortalidad que los pacientes con HAP idiopática23,24, por lo que se recomienda su referencia a centros expertos [I, C]. No se aconseja el empleo de anticoagulantes [III, C] ni de betabloqueantes25. La hipertensión portopulmonar es un factor de riesgo mayor para el trasplante hepático26, por lo que debe descartarse mediante ETT en todos los candidatos a trasplante [I, B] y confirmarse con estudio hemodinámico. Si la PAPm es<35mmHg puede considerarse el trasplante26 [IIb, C]. Si es≥35mmHg se aconseja instaurar terapia específica y reevaluar a los 3 meses. Si la PAP persiste elevada a pesar del tratamiento, está contraindicado el trasplante hepático [III, C].
Infección por VIHLa ETT para detectar HAP debe realizarse en casos de disnea inexplicable [III, C]. No se recomienda la anticoagulación por el riesgo de sangrado y las posibles interacciones farmacológicas [III, C]. Deben tenerse en cuenta las interacciones de los IPDE5 con algunos antirretrovirales.
Enfermedad venooclusiva pulmonar y hemangiomatosis capilar pulmonarLa EVOP y la HCP comparten características clínicas, patológicas y genéticas, así como el tratamiento27. La EVOP puede asociarse a esclerosis sistémica, infección por VIH o fármacos. La forma familiar está causada por la mutación bialélica del gen EIF2AK428.
El diagnóstico se establece por criterios clínicos, exploración física, broncoscopia y radiología [I, C], o bien la identificación de la mutación del gen EIF2AK4 [I, B].
Los agentes vasodilatadores pueden producir edema pulmonar en la EVOP/HCP. El trasplante pulmonar es el tratamiento de elección, por lo que se debe referir a los pacientes a una unidad de trasplante pulmonar tras el diagnóstico [I, C].
Situaciones especialesEmbarazo y control de la natalidad: debido al elevado riesgo de mortalidad, las pacientes con HAP deben evitar el embarazo [I, C]. Se aconseja combinar 2 métodos anticonceptivos. Los progestágenos son preferibles a los estrógenos. En caso de embarazo, debe informarse del riesgo y proponer la interrupción del mismo. Las pacientes que decidan asumir el riesgo y continuar el embarazo deben ser controladas estrechamente en un centro con unidades expertas en HP y en embarazo de riesgo.
Cirugía: constituye una situación de elevado riesgo de morbimortalidad, especialmente si es no programada29, por lo que se recomienda su realización en centros de referencia en HP. Es preferible la anestesia epidural a la general29.
Insuficiencia cardiaca derecha: los diuréticos producen beneficio sintomático. En situaciones que requieran ingreso en la UCI se debe optimizar el balance hídrico con diuréticos iv, reducir la sobrecarga del ventrículo derecho (habitualmente con prostanoides iv), mejorar el gasto cardiaco (preferiblemente con dobutamina) y evitar la intubación, que frecuentemente produce colapso hemodinámico. El uso de ECMO y otros dispositivos debería considerarse en pacientes seleccionados30.
Hipertensión pulmonar debida a cardiopatía izquierdaLa HP asociada a cardiopatía izquierda es de tipo poscapilar. Se distinguen 2 tipos: HP poscapilar aislada e HP combinada poscapilar y precapilar, de acuerdo con los valores del gradiente de presión diastólica y la resistencia vascular pulmonar (tabla 9).
Clasificación hemodinámica de la hipertensión pulmonar poscapilar
| Definición | Características |
|---|---|
| HP poscapilar | PAPm≥25mmHg PAWP>15mmHg |
| HP poscapilar aislada | GPD<7mmHg y/o RVP≤3UW |
| HP combinada poscapilar y precapilar | GPD≥7mmHg y/o RVP>3UW |
GPD: gradiente de presión diastólica (PAP diastólica−PAWP); HP: hipertensión pulmonar; PAPm: presión arterial pulmonar media; PAWP: presión de oclusión de la arteria pulmonar; RVP: resistencia vascular pulmonar; UW: unidades Wood (mmHg/L/min).
El diagnóstico diferencial entre HAP e HP del grupo 2 puede ser complejo, especialmente en los pacientes con HP e insuficiencia cardiaca con fracción de eyección preservada. Se prestará atención a las características indicadas en la tabla 1031.
Datos indicativos de insuficiencia cardiaca con fracción de eyección preservada
| Edad>65 años |
| Factores de riesgo cardiovascular: diabetes mellitus, dislipidemia o hipertensión sistémica |
| Enfermedad coronaria |
| Fibrilación auricular |
| Ecocardiografía: mayor dilatación de la aurícula izquierda que de la derecha, hipertrofia del ventrículo izquierdo, abombamiento del septo interauricular hacia la aurícula derecha, disfunción diastólica en el doppler del flujo mitral |
| ECG: presencia de hipertrofia de ventrículo izquierdo y ondas Q |
El abordaje se basa en optimizar el tratamiento de la insuficiencia cardiaca [I, B]. Los pacientes con HP combinada poscapilar y precapilar grave deben ser remitidos a centros expertos para ser incluidos en ensayos clínicos y/o recibir tratamiento individualizado [IIa, C]. Se desaconseja la utilización de fármacos indicados para la HAP [III, C].
Hipertensión pulmonar debida a enfermedad respiratoriaLas enfermedades pulmonares que más comúnmente se asocian a HP son la EPOC, las neumopatías intersticiales y la combinación de fibrosis pulmonar y enfisema. En la tabla 11 se muestra la clasificación hemodinámica de la HP en este grupo. La HP habitualmente es de grado leve o moderado32. La HP severa se observa con mayor frecuencia en la combinación de fibrosis pulmonar y enfisema y comúnmente se asocia a una capacidad de difusión de CO desproporcionadamente reducida y una PaCO2 baja33.
Clasificación hemodinámica de la hipertensión pulmonar asociada a enfermedades respiratorias
| Terminología | Características hemodinámicas |
|---|---|
| EPOC/FPI/CFPE sin HP | PAPm<25mmHg |
| EPOC/FPI/CFPE con HP | PAPm≥25mmHg |
| EPOC/FPI/CFPE con HP grave | PAPm>35mmHg o PAPm≥25mmHg en presencia de bajo gasto cardiaco (IC<2,5L/min, no explicable por otras causas) |
CFPE: combinación de fibrosis pulmonar y enfisema; EPOC: enfermedad pulmonar obstructiva crónica; FPI: fibrosis pulmonar idiopática; HP: hipertensión pulmonar; IC: índice cardiaco; PAPm: presión arterial pulmonar media.
La ETT es la exploración de elección para la detección de HP [I, C], aunque su precisión en pacientes con enfermedad respiratoria avanzada es baja. Está indicada si se sospecha HP significativa o para descartar cardiopatía izquierda.
El diagnóstico definitivo de HP se establece mediante CCD. Sus indicaciones son: 1) diagnóstico adecuado o exclusión de HP en candidatos a tratamientos quirúrgicos (trasplante, reducción de volumen pulmonar); 2) sospecha de HAP o HPTEC concomitantes; 3) episodios repetidos de insuficiencia cardiaca derecha, y 4) ETT no concluyente en casos con alta sospecha de HP34.
El tratamiento de elección en los pacientes con EPOC e HP hipoxémicos es la oxigenoterapia continua domiciliaria [I, C]. En las enfermedades intersticiales el papel de la oxigenoterapia es menos claro.
No se recomiendan los vasodilatadores convencionales ni los fármacos específicos de HAP en la EPOC con HP leve-moderada [III, C]2,35. En la fibrosis pulmonar idiopática está contraindicado el uso de ambrisentán y de riociguat [III, A]. Los pacientes con enfermedad respiratoria e HP severa deberían ser remitidos a un centro especializado en ambos procesos para un tratamiento individualizado [I, C].
Hipertensión pulmonar tromboembólica crónicaEl diagnóstico de HPTEC se establece por la presencia de trombosis pulmonar e HP precapilar, tras más de 3 meses de tratamiento anticoagulante correcto.
El algoritmo diagnóstico de la HPTEC (fig. 3) tiene 2 componentes: el diagnóstico hemodinámico mediante CCD y la localización de las lesiones trombóticas mediante técnicas de imagen (angio-TC y angiografía pulmonar selectiva de sustracción digital).

Existen 3 opciones terapéuticas en la HPTEC (fig. 4):
- 1.
Tratamiento quirúrgico
La endarterectomía pulmonar (EAP) es el tratamiento de elección [I, C]. Esta intervención puede lograr la curación de la HPTEC y es apropiada para más del 60% de los casos.Todo paciente diagnosticado de HPTEC debería ser evaluado para la posible indicación de EAP en un centro con experiencia en esta cirugía, por parte de un equipo multidisciplinar que incluya un cirujano especializado [I, C]. En España hay 2 centros acreditados36–38.
- 2.
Tratamiento médico
Los pacientes con HPTEC deben recibir anticoagulación indefinida, incluso tras la EAP [I, C]. Se recomienda el uso de antagonistas de la vitamina K, ya que no hay evidencia con el uso de los nuevos anticoagulantes orales. Actualmente, el único fármaco específico con indicación para la HPTEC es riociguat39 [I, B]. También se han demostrado efectos beneficiosos con macitentán40 y de forma parcial con bosentán41. El tratamiento farmacológico está indicado en pacientes en quienes un comité multidisciplinar experto en EAP ha desestimado la cirugía y en la HP persistente tras EAP [I, B].
- 3.
Angioplastia pulmonar
La angioplastia pulmonar con balón es un procedimiento de desarrollo reciente que ha proporcionado buenos resultados en pacientes con lesiones obstructivas no accesibles mediante EAP42–44, aunque la evidencia disponible es todavía escasa45. Este procedimiento solo debe realizarse en centros con amplia experiencia en HPTEC, una vez se haya desestimado la EAP2.

Este grupo incluye procesos de etiopatogenia variada: enfermedades hematológicas, enfermedades sistémicas, trastornos metabólicos y un grupo misceláneo de procesos (tabla 2). Su diagnóstico es difícil, por lo que es aconsejable su manejo en centros con experiencia en HP. En la actualidad no hay aprobado ningún tratamiento específico para este grupo.
Organización asistencialLas formas no secundarias de HP (grupos 1, 4 y 5) (tabla 2) son enfermedades poco prevalentes y graves, que requieren procedimientos complejos para su diagnóstico y tratamiento. Existe un amplio consenso en que los pacientes con enfermedades de estas características deben ser atendidos en unidades de referencia con experiencia en la enfermedad2,46. En el año 2008, la Sociedad Española de Neumología y Cirugía Torácica y la Sociedad Española de Cardiología elaboraron un documento de consenso en el que se proponía para España una organización asistencial para la atención de los pacientes con HP basada en unidades expertas en HP que interactúan en red con centros de ámbito local46. Los criterios que deben cumplir las unidades expertas en HP establecidos en la guía clínica de la European Society of Cardiology-European Respiratory Society2 se muestran en la tabla 12. Desde el año 2015, en España existen 3 CSUR (centros, servicios o unidades de referencia) de HP compleja de ámbito nacional, designados por el Ministerio de Sanidad.
Recomendaciones para las unidades expertas en hipertensión pulmonar
| Disponibilidad de un equipo multidisciplinar de profesionales [I, C] |
| Seguimiento de>50 pacientes con HAP o HPTEC (idealmente>200) [IIa, C] |
| Recibir>24 casos nuevos al año con el diagnóstico de HAP o HPTEC [IIa, C] |
| Efectuar>20 cateterismos cardiacos derechos con prueba vasodilatadora al año [IIa, C] |
HAP: hipertensión arterial pulmonar; HPTEC: hipertensión pulmonar tromboembólica crónica.
[Clase de recomendación, nivel de evidencia].
Dada la estructura organizativa de la sanidad española, la organización asistencial de la HP en España debería configurarse como una red de redes, con unidades expertas en HP de ámbito autonómico, que interactúan con centros asociados dentro de la propia comunidad autónoma, y los CSUR de ámbito nacional, que disponen de programas de EAP y capacidad para atender a los pacientes y las situaciones de mayor complejidad.
Las unidades expertas en HP deben tener establecidos circuitos de consulta y derivación para enfermedades o situaciones clínicas concretas: HPTEC (EAP, angioplastia pulmonar), trasplante pulmonar, hipertensión portopulmonar, cardiopatías congénitas, enfermedades del tejido conectivo, estudio genético, cirugía electiva y atención de la paciente embarazada.
Conflicto de interesesEl Dr. Barberà ha recibido honorarios de Actelion, Bayer, GlaxoSmithKline, Merck Sharp & Dohme y Pfizer, y ha recibido financiación de Actelion, Bayer, GlaxoSmithKline, y Pfizer, no relacionados con el trabajo presentado. La Dra. Blanco ha recibido honorarios de Merck Sharp & Dohme, no relacionados con el trabajo presentado. La Dra. Otero Candelera ha recibido honorarios Actelion Bayer, Rovi, Leo Pharma y Merck Sharp & Dohme, y ha recibido financiaciónde Bayer y Leo Pharma, no relacionadas con el trabajo presentado. La Dra. López-Reyes ha recibido honorarios de Actelion, y finaciación de GlaxoSmithKline, Ferrer y Actelion, no relacionados con el trabajo presentado. La Dra. Otero ha recibido honorarios de Actelion, Bayer, Glaxo-SmithKline y Ferrer, no relacionados con el trabajo presentado. El Dr. Pérez-Peñate ha recibido honorarios de Actelion, Bayer y Merck Sharp & Dohme, no relacionados con el trabajo presentado. El Dr. Sala no tiene conflictos que declarar. La Dra. Escribano ha recibido honorarios de Actelion, Bayer, GlaxoSmithKline, y Merck Sharp & Dohme, y ha recibido financiación de Actelion, Bayer, GlaxoSmithKline y Ferrer, no relacionados con el trabajo presentado.
Los miembros del grupo de trabajo agradecen la revisión del manuscrito y los comentarios efectuados por A. Ballaz, J. de Miguel, J. Guerra y G. Juan.







