


El síndrome de Hermansky-Pudlak (SHP) es una enfermedad autosómica recesiva que comúnmente se presenta en latinos de ascendencia puertorriqueña. Presentamos 2 casos clínicos de fibrosis pulmonar familiar en 2 hermanas mexicanas con SHP. La fibrosis pulmonar se confirmó por biopsia en una paciente. Esta comunicación demuestra que el SHP puede aparecer en población mexicana.
Hermansky-Pudlak syndrome is an autosomal recessive disorder commonly found in individuals of Puerto Rican ancestry. We present 2 cases of familial pulmonary fibrosis in 2 Mexican sisters with Hermansky-Pudlak syndrome. Pulmonary fibrosis was biopsy-proven in 1 of the patients. This report shows that Hermansky-Pudlak syndrome may occur in individuals of Mexican ancestry.
El síndrome de Hermansky-Pudlak (SHP) es una infrecuente enfermedad autosómica recesiva, que se caracteriza por albinismo, diátesis hemorrágica y fibrosis pulmonar. La mayoría de los casos comunicados en la literatura médica corresponde a personas de ascendencia puertorriqueña. Hasta ahora no se ha descrito el SHP con fibrosis pulmonar familiar en personas mexicanas. Por lo tanto, consideramos de suma relevancia la difusión del caso de 2 hermanas mexicanas con esta asociación.
Observación clínicaCaso 1Mujer de 60 años, albina, con epistaxis y púrpura de 2 años de evolución. Un año antes de acudir a consulta había comenzado a presentar disnea al ejercicio y tos no productiva. Las pruebas de función pulmonar mostraron un grave patrón restrictivo, con capacidad vital forzada del 42%, volumen espiratorio forzado en el primer segundo del 50% y capacidad de difusión de monóxido de carbono del 60% del predicho. La tomografía computarizada de alta resolución (figura 1A) mostró una imagen discreta y difusa de vidrio deslustrado, así como opacidades reticulares en parche sin panal de abeja. La biopsia de pulmón a cielo abierto demostró neumonitis intersticial fibrosante, de predominio subpleural, panal de abeja microscópico y focos de fibroblastos. Había áreas de pulmón normal adyacentes a las zonas de pulmón con fibrosis. Además se observaron áreas de neumonía organizada en parche, así como proliferación y vacuolización de neumocitos tipo 2 con zonas de bronquiolitis constrictiva y metaplasia bronquiolar o lambertosis. Estos hallazgos eran indicativos de neumonía intersticial usual, pero con características especiales (figura 2). Dada la presencia de albinismo, se realizó una aspiración de médula ósea que mostró megacariocitos llenos de gránulos densos y cuerpos de lipofuscina, lo que permitió confirmar el diagnóstico de SHP. La paciente presentó un curso agresivo y progresivo de la disnea, y murió un mes después.
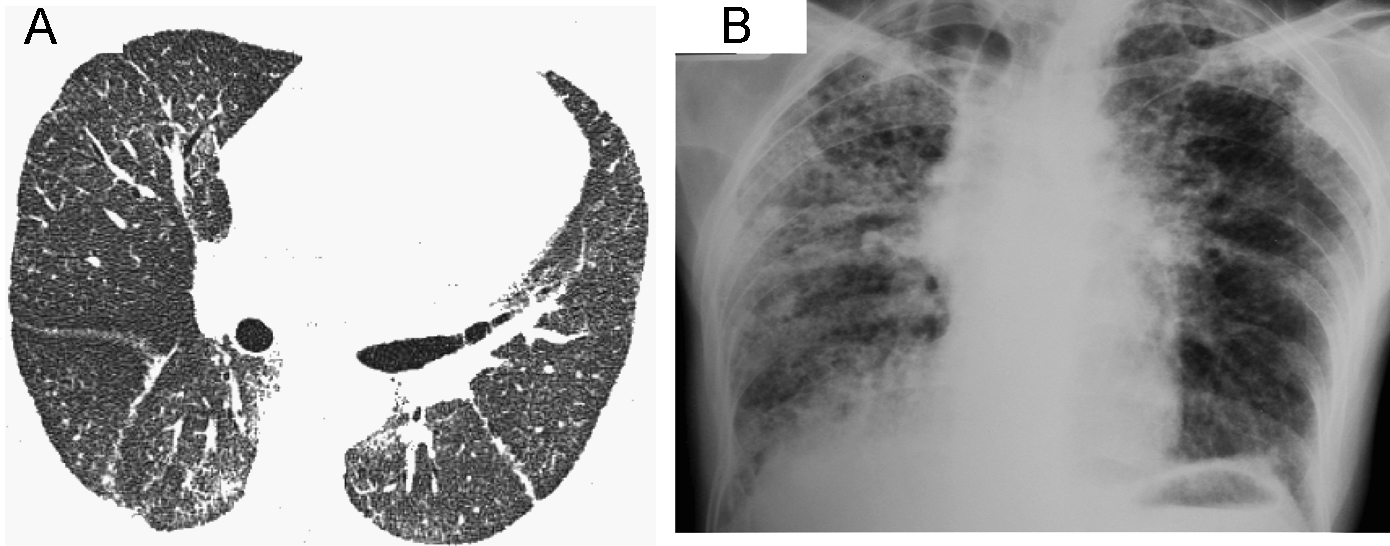
Figura 1. La tomografía computarizada de alta resolución (A) muestra una imagen discreta y difusa de vidrio deslustrado, así como opacidades reticulares periféricas y en parche. No se aprecia panal de abeja. La radiografía de tórax (B) demostró disminución de los volúmenes pulmonares e infiltrados reticulonodulares de predominio apical, así como panal de abeja periférico y bibasal con ensanchamiento de la arteria pulmonar.
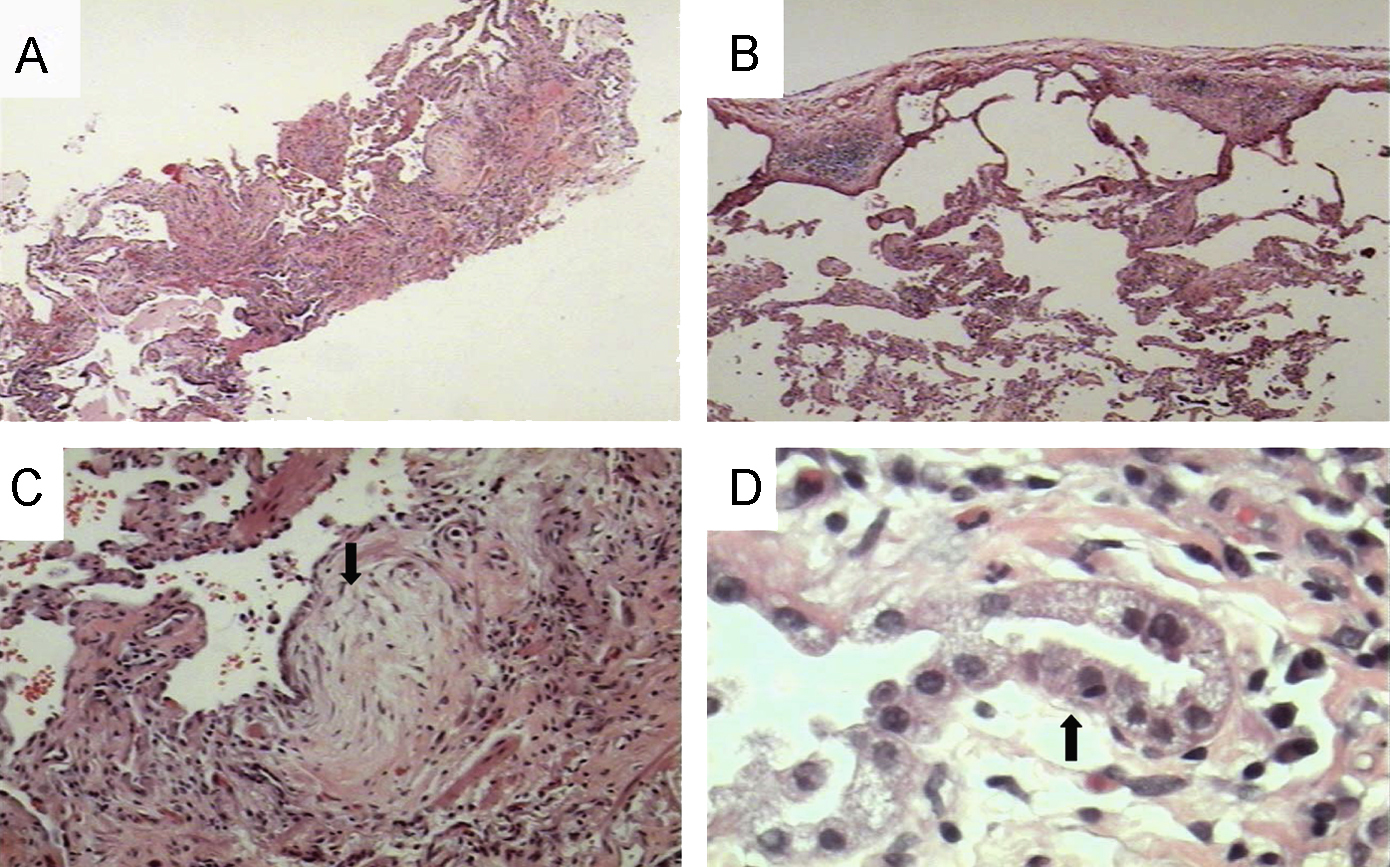
Figura 2. A y B: vistas panorámicas donde se aprecia la afección predominantemente periférica. (Hematoxilina-eosina, aumento original ×1 y ×10.) C: se aprecia un foco de miofibroblastos, indicado por la flecha. (Hematoxilina-eosina, aumento original ×10.) D: gran hipertrofia y vacuolización de neumocitos tipo 2 (flecha). (Hematoxilina-eosina, aumento original ×40.)
Caso 2Mujer de 56 años, con albinismo oculocutáneo, hermana de la paciente anterior, a quien se examinó en nuestra institución 10 años antes de tratar al caso 1. Acudió a consulta con una historia de 10 meses de evolución de disnea al ejercicio, tos seca y dolor torácico. Desde hacía 3 años presentaba diátesis hemorrágica caracterizada por epistaxis, hematomas y gingivorragia. Se le realizó una espirometría que reveló un grave patrón restrictivo, con capacidad vital forzada del 31% y volumen espiratorio forzado en el primer segundo del 37% del predicho. Los estudios de laboratorio hematológicos demostraron que tanto las plaquetas como los tiempos de coagulación estaban dentro de los límites normales. La radiografía de tórax demostró disminución de los volúmenes pulmonares e infiltrados reticulonodulares de predominio apical, así como panal de abeja periférico y bibasal con ensanchamiento de la arteria pulmonar (figura 1B). Debido a lo avanzado del daño pulmonar no fue posible realizar biopsia pulmonar a cielo abierto. Se le administró tratamiento con oxígeno, el curso fue progresivo y la paciente murió un mes después de acudir a consulta.
Diez años más tarde, al examinar a su hermana, el caso 1, en el que se comprobó la presencia de SHP por biopsia de médula ósea, reevaluamos el caso 2, que presentaba los mismos síntomas tanto hematológicos como pulmonares, así como los rasgos fenotípicos de albinismo, lo que constituye la tríada del SHP, que se compone de albinismo, diátesis hemorrágica y fibrosis pulmonar1.
DiscusiónEl SHP se describió por primera vez en 1959, cuando los Dres. Hermansky y Pudlak comunicaron la presencia, en pacientes albinos, de un síndrome caracterizado por diátesis hemorrágica y fibrosis pulmonar2. En Puerto Rico el SHP se encuentra en 5 de cada 6 albinos3. Se han identificado al menos 8 subtipos de SHP en personas de diferentes etnias (Puerto Rico, Japón y Europa). Los subtipos 1 y 4 son los que se asocian a una fibrosis pulmonar más grave4. En México el SHP es extremadamente infrecuente: sólo se han comunicado 3 casos en la literatura médica1,5. La fibrosis pulmonar que desarrollaron las pacientes estudiadas fue muy intensa, lo que indica que posiblemente presentaran el subtipo 1 o 4. Sin embargo, no tenían ascendencia puertorriqueña y era descendientes de mestizos mexicanos. Esto indica que es posible que el síndrome fuera consecuencia de una mutación de novo; de hecho, en la literatura médica se ha comunicado que un 50% de los pacientes de origen no puertorriqueño pueden presentar mutaciones de novo4.
El SHP es un trastorno autosómico recesivo, consecuencia de una formación y un transporte anormales de vesículas intracelulares en melanosomas, plaquetas y lisosomas, lo que da lugar a la acumulación de cuerpos ceroides de lipofuscina6,7. La patogenia de la fibrosis pulmonar en el SHP parece estar asociada a la acumulación de estos cuerpos ceroides de liposfuscina en el interior de los neumocitos tipo 2, unida a la acumulación de surfactante y otras moléculas en macrófagos. Asimismo, en modelos animales de SHP se ha descrito la deficiencia de secreción de surfactante, lo que conduce a la acumulación de cuerpos lamelares gigantes en neumocitos tipo 28. Esta acumulación anormal de proteínas en el compartimiento endosómico lleva a la reparación aberrante y a la fibrosis en respuesta a una lesión9,10. Es interesante observar que la patogenia del SHP guarda cierta similitud con las hipótesis propuestas para la patogenia de la fibrosis pulmonar familiar, donde algunas proteínas como el surfactante C no funcionan correctamente debido a un plegamiento anormal de las mismas11,12. Esto da lugar a un estado de estrés del retículo endoplásmico, lo que genera una respuesta de reparación anormal y, por lo tanto, fibrosis13,14.
La fibrosis pulmonar secundaria al SHP tiene características únicas que la distinguen de la neumonía intersticial usual, que por regla general se presenta en pacientes con fibrosis pulmonar idiopática (FPI). Estas características son: degeneración vacuolar de los neumocitos tipo 2, bronquiolitis constrictiva y panal de abeja microscópico que no necesariamente se encuentra en la periferia, como sucede en pacientes con FPI9,15. En el caso 1 se sospechó la presencia de SHP no sólo por el albinismo, sino sobre todo porque la tomografía computarizada de alta resolución no era típica de la FPI asociada a neumonía intersticial usual, ya que mostraba imagen de vidrio deslustrado y no había panal de abeja macroscópico. Más aún, en la histopatología se observaban 2 de las 3 características patológicas del SHP: bronquiolitis constrictiva y degeneración vacuolar de los neumocitos tipo 2. Cabe mencionar que, aunque estos cambios histopatológicos se parecen a los de la neumonía intersticial usual, el curso de nuestras pacientes fue mucho más agresivo que el de los afectados por FPI asociada a neumonía intersticial usual, que, como se sabe, tienen un pronóstico de 3 años a partir del diagnóstico16.
En conclusión, en nuestro conocimiento, éste es el primer caso comunicado de SHP asociado a fibrosis pulmonar familiar en población mexicana. Si bien el SHP está bien reconocido en pacientes puertorriqueños, hay que tener en mente este diagnóstico en pacientes mexicanos/mestizos.







