



Las mutaciones del gen que codifica el receptor 2 de las proteínas morfogénicas del hueso (BMPR2) contribuyen a la patogénesis de la hipertensión arterial pulmonar en sus formas familiar (HAPF) e idiopática.
MétodoCon el objetivo de profundizar en el conocimiento de dichos factores genéticos en nuestro medio, se estudió el gen BMPR2 en 17 pacientes con hipertensión arterial pulmonar, 8 con HAPF y 9 con hipertensión arterial idiopática esporádica. Adicionalmente, se analizó si la presencia de mutaciones del gen BMPR2 se asociaba a cambios en la capacidad de difusión del CO a fin de evaluar el interés de esta medición en el diagnóstico preclínico.
ResultadosSe detectaron las mutaciones R491Q y R211X en 2 pacientes con HAPF (prevalencia 25%), y la mutación R332X en un caso de hipertensión arterial idiopática (prevalencia 11%). El estudio familiar del paciente con la mutación R491Q demostró la presencia de la misma en 14 de los 28 sujetos estudiados, de los cuales 5 presentaban la enfermedad (penetrancia 36%). En dicha familia se observó un descenso de la relación en la capacidad de difusión del CO/volumen alveolar en los familiares asintomáticos que expresaban la mutación, comparado con los que no la expresaban (88±5% y 104±9% del valor de referencia, respectivamente; p<0,01).
ConclusiónConcluimos que la frecuencia de mutaciones del gen BMPR2 en los pacientes con HAPF estudiados es inferior a la descrita previamente. El descenso del volumen alveolar observado en portadores de la mutación asintomáticos sugiere cierto grado de alteración vascular pulmonar, por lo que su medición podría ser útil en el estudio familiar de la HAPF.
Mutations of the gene that code bone morphogenic protein type 2 receptor (BMPR2) are involved in the pathogenesis of pulmonary arterial hypertension (PAH), both in its familial (FPAH) and its idiopathic (IPAH) forms.
MethodWith the aim of increasing the knowledge of these genetic factors in our area, the BMPR2 gene was studied in 17 patients with PAH, 8 with FPAH and 9 with sporadic IPAH. Additionally, a study was made to see whether the presence of BMPR2 mutations was associated with changes in the CO diffusing CO (DLCO) with the aim of evaluating the interest in this measurement in the pre-clinical diagnosis.
ResultsR491Q y R211X mutations were detected in 2 patients with FPAH (prevalence, 25%), and the R332X mutation in one case of IPAH (prevalence, 11%). The familial study of the patient with the R491Q mutation, 14 of the 28 subjects studied had the mutation, and 4 had the diseases (penetration, 36%). A decrease in the DLCO/alveolar volume (KCO) ratio was observed in asymptomatic family members who expressed the mutation, compared to those who did not express it (88±5% and 104±9% of the reference value, respectively; P<0.01).
ConclusionWe conclude that the frequency of mutations in the BMPR2 gene in the patients studied with FPAH is lower than was previously described. The decrease in the KCO observed in asymptomatic carriers of the mutation suggests a certain level of pulmonary vascular changes, therefore its measurement could be useful in the familial study of FPAH.
La hipertensión arterial pulmonar (HAP) es una enfermedad de etiología desconocida, caracterizada por el aumento de presión en la arteria pulmonar1. Desde que la HAP fue bien caracterizada en la década de los 80, se sabe que un porcentaje significativo de casos presentan antecedentes familiares de la enfermedad2, lo que se conoce como HAP hereditaria o familiar (HAPF). Este hecho condujo a la búsqueda de factores genéticos que pudieran explicar el origen de la misma. En el año 2000, Deng et al3 localizaron por ligamiento genético la región 2q33 como región candidata y, posteriormente, se describieron mutaciones en el gen BMPR2 como causa de la enfermedad (International Primary Pulmonary Hipertensión Consortium)4. El gen BMPR2 codifica el receptor 2 de las proteínas morfogénicas del hueso, que pertenecen a la superfamilia del factor de crecimiento transformante β. Aproximadamente, un 70% de los pacientes con HAPF presentan mutaciones en dicho gen, del cual no hay mutaciones recurrentes, y del que hasta la fecha se han identificado casi 300 mutaciones distintas5. También, se han descrito mutaciones del gen BMPR2 en casos de HAP idiopática (HAPI) esporádica6,7, aunque con una menor frecuencia que en la HAPF, en la HAP asociada al consumo de anorexígenos8 y en la asociada a cardiopatías congénitas9. Se desconoce si la prevalencia de estas mutaciones varía en relación con el origen geográfico o el grupo étnico. La enfermedad se hereda siguiendo un patrón de herencia autosómica dominante con penetrancia reducida. Tan solo del 10 al 20% de los portadores de la mutación expresan fenotípicamente la enfermedad.
Hasta la fecha, no se han publicado estudios del gen BMPR2 realizados en pacientes con HAPF en España. Solamente existe un estudio en población española realizado en 8 casos con HAPI esporádica10.
En la actualidad no se dispone de ningún biomarcador o medida funcional que permita detectar precozmente el riesgo de desarrollar HAP en los portadores asintomáticos de la mutación del gen BMPR2. Sin embargo, existe la sospecha de que el lecho vascular pulmonar de los portadores sanos de la mutación es anómalo, ya que dichos sujetos desarrollan hipertensión pulmonar durante el esfuerzo11. La capacidad de difusión del monóxido de carbono (DLCO) está característicamente disminuida en los pacientes con HAP. Dado que este parámetro puede estar alterado por la reducción del lecho vascular pulmonar, hemos planteado la hipótesis de que la DLCO podría estar también reducida en los portadores sanos de la mutación del gen BMPR2.
Con el objetivo de profundizar en el conocimiento de los factores genéticos asociados al desarrollo de HAP en nuestro medio, se estudió el gen BMPR2 en una serie de 17 pacientes, 8 con HAP familiar y 9 con HAPI esporádica. Adicionalmente, se analizó si la presencia de mutaciones del gen BMPR2 se asociaba a cambios en la DLCO, a fin de evaluar el posible interés de dicha medición en el diagnóstico preclínico de la enfermedad.
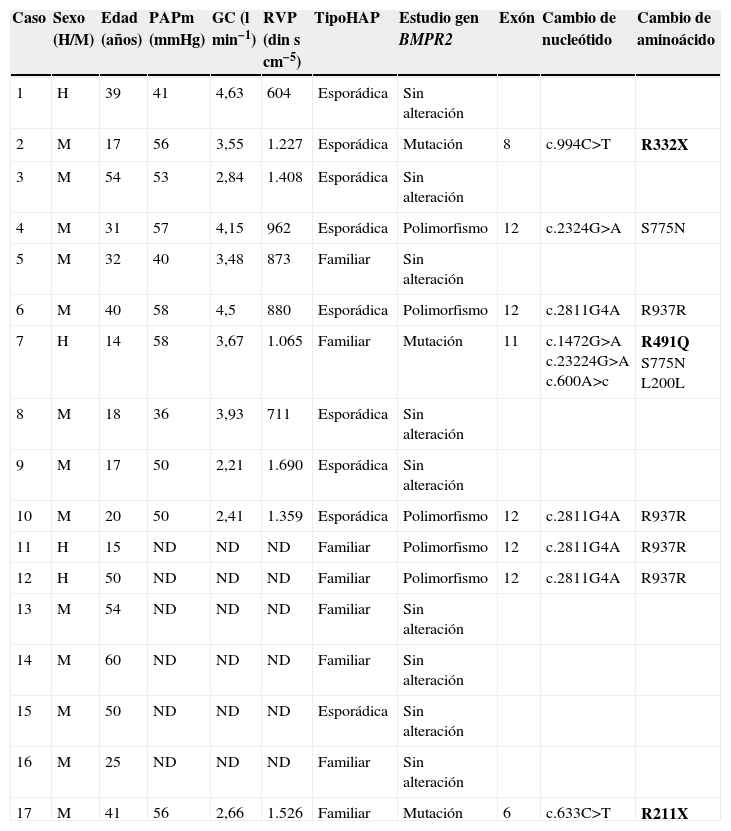
MétodosSujetos de estudioSe han estudiado consecutivamente 17 casos independientes de pacientes diagnosticados de HAP, 8 con HAPF y 9 casos con HAPI esporádica (tabla 1).
Características demográficas, hemodinámicas y resultados del estudio del gen BMPR2
| Caso | Sexo (H/M) | Edad (años) | PAPm (mmHg) | GC (lmin−1) | RVP (dinscm−5) | TipoHAP | Estudio gen BMPR2 | Exón | Cambio de nucleótido | Cambio de aminoácido |
| 1 | H | 39 | 41 | 4,63 | 604 | Esporádica | Sin alteración | |||
| 2 | M | 17 | 56 | 3,55 | 1.227 | Esporádica | Mutación | 8 | c.994C>T | R332X |
| 3 | M | 54 | 53 | 2,84 | 1.408 | Esporádica | Sin alteración | |||
| 4 | M | 31 | 57 | 4,15 | 962 | Esporádica | Polimorfismo | 12 | c.2324G>A | S775N |
| 5 | M | 32 | 40 | 3,48 | 873 | Familiar | Sin alteración | |||
| 6 | M | 40 | 58 | 4,5 | 880 | Esporádica | Polimorfismo | 12 | c.2811G4A | R937R |
| 7 | H | 14 | 58 | 3,67 | 1.065 | Familiar | Mutación | 11 | c.1472G>A c.23224G>A c.600A>c | R491Q S775N L200L |
| 8 | M | 18 | 36 | 3,93 | 711 | Esporádica | Sin alteración | |||
| 9 | M | 17 | 50 | 2,21 | 1.690 | Esporádica | Sin alteración | |||
| 10 | M | 20 | 50 | 2,41 | 1.359 | Esporádica | Polimorfismo | 12 | c.2811G4A | R937R |
| 11 | H | 15 | ND | ND | ND | Familiar | Polimorfismo | 12 | c.2811G4A | R937R |
| 12 | H | 50 | ND | ND | ND | Familiar | Polimorfismo | 12 | c.2811G4A | R937R |
| 13 | M | 54 | ND | ND | ND | Familiar | Sin alteración | |||
| 14 | M | 60 | ND | ND | ND | Familiar | Sin alteración | |||
| 15 | M | 50 | ND | ND | ND | Esporádica | Sin alteración | |||
| 16 | M | 25 | ND | ND | ND | Familiar | Sin alteración | |||
| 17 | M | 41 | 56 | 2,66 | 1.526 | Familiar | Mutación | 6 | c.633C>T | R211X |
BMPR2: receptor 2 de las proteínas morfogénicas del hueso; GC: gasto cardíaco; HAP: hipertensión arterial pulmonar; ND: no disponible; PAPm: presión arterial pulmonar media; RVP: resistencia vascular pulmonar.
También se evaluaron 3 casos independientes de familiares sanos de pacientes fallecidos con sospecha de HAPF remitidos para estudio genético. Adicionalmente, se han analizado 50 individuos sanos no relacionados con la enfermedad como controles para el estudio del polimorfismo S775N.
El diagnóstico de HAP se estableció mediante estudio hemodinámico pulmonar en todos los casos, y fue definida por la presencia de presión arterial pulmonar media ≥25mmHg en reposo y presión de oclusión arterial pulmonar ≤15mmHg, en ausencia de otras causas asociadas1. Se consideraron los siguientes criterios para establecer el diagnóstico de HAPF: 1) pacientes con familiares diagnosticados de HAP mediante estudio hemodinámico pulmonar, o 2) antecedentes familiares de insuficiencia cardiaca derecha aislada o muerte súbita no justificada por otro origen. Las características hemodinámicas de los pacientes se obtuvieron de los registros de la historia clínica o fueron proporcionadas por los médicos que remitieron a los pacientes a nuestro centro.
Estudio molecular del gen BMPR2El método de estudio ha sido la amplificación de los 13 exones por PCR según primers descritos por Deng et al3 modificados y valoración mediante Single Strand Conformation Polymorphism (SSCPs) de los fragmentos de migración anómalos y posterior secuenciación de los mismos en un analizador ABI3100.
Solo se estudió la zona codificante y 50 pares de bases flanqueantes intrón-exón. No se analizaron cambios en otras regiones.
Caracterización clínica y funcional de familias con casos de hipertensión arterial pulmonar familiarEn familiares de 2 pacientes con HAP familiar se realizaron, además del análisis genético, estudios clínicos, examen funcional respiratorio y ecocardiografía Doppler. Las dos familias eran de origen español. Se evaluaron 28 miembros de la familia del caso 7, y 5 de la familia del caso 5. Todos los sujetos manifestaron su consentimiento por escrito tras ser informados de las características y objetivos del estudio. El estudio clínico consistió en la anamnesis detallada, preguntando específicamente por sintomatología cardiaca o respiratoria, exploración física, radiografía de tórax, electrocardiograma y ecocardiografía. El estudio funcional respiratorio, incluyó espirometría forzada y medición de DLCO.
La espirometría forzada y la medición de DLCO (Master Screen; Jaeger, Wüerzburg, Alemania) fueron realizadas de acuerdo con las recomendaciones de la Sociedad Española de Neumología y Cirugía Torácica12. El estudio ecocardiográfico se realizó mediante ecocardiograma transtorácico (Sonos 5500, Phillips, Holanda) con transductores de 2,5–3,5MHz. Se registraron las medidas de las cavidades en modo M y de los volúmenes ventriculares por 2D, con examen Doppler de las velocidades de los flujos transvalvulares y la determinación de la velocidad de regurgitación tricuspídea desde los distintos planos de estudio.
Con posterioridad, se ha efectuado seguimiento clínico periódico a aquellos sujetos que resultaron positivos para la mutación.
Análisis estadísticoLos datos se expresan con media±SD para las variables cuantitativas y como porcentaje sobre los valores absolutos para las cualitativas. Las medias de los dos grupos se compararon con la prueba t de Student para medidas independientes. Se consideraron significativas las diferencias cuyo valor de p asociado a la prueba de contraste fue <0,05.
ResultadosLos resultados de los estudios hemodinámico y genético se muestran en la tabla 1.
Estudio genético del gen BMPR2Se han detectado 10 cambios en la secuencia del gen BMPR2 respecto a la secuencia normal, aunque solo 3 de ellos corresponden a mutaciones previamente descritas como causantes de la enfermedad. La primera (R332X) es una mutación que crea un codon stop y, por lo tanto, una proteína truncada (fig. 1) descrita en el año 2000 por Thomson et al6 como causante de la enfermedad. En nuestra serie se detectó en uno de los casos del grupo de HAPI esporádica (caso 2), lo que da lugar a una frecuencia de mutaciones del 11,1% en dicho grupo.
 Caso 7, exón 11 c.1472G>A, p.R491Q. B) Caso 2, exón 7 c.994C>T; p.R332X. C) Caso 17, exón 6 c.633C>T; p.R211X.")
Secuencias parciales del gen BMPR2. En la parte superior, la secuencia normal y, en la inferior, la secuencia parcial del exón correspondiente mostrando la mutación. El círculo indica el cambio.
A) Caso 7, exón 11 c.1472G>A, p.R491Q.
B) Caso 2, exón 7 c.994C>T; p.R332X.
C) Caso 17, exón 6 c.633C>T; p.R211X.
La segunda mutación (R491Q), detectada en el caso 7 (grupo HAPF), es una mutación tipo missense descrita en el año 2000 por Deng et al3 (fig. 1). Por último, la tercera mutación (R211X), detectada en el caso 17 (grupo HAPF), da lugar, al igual que la primera, a un codon stop y, por lo tanto, a una proteína truncada (fig. 1)13. Dado que se han identificado 2 mutaciones en un total de 8 casos con HAPF, la frecuencia de mutaciones en estos casos es del 25%.
Para la mutación R491Q se ha podido estudiar a un total de 28 miembros de la familia, en los cuales se ha detectado la mutación en 14. De estos, 5 casos presentaron la enfermedad, por lo que la penetrancia en esta familia es del 35,7%.
Por otra parte, se han detectado varios polimorfismos sin responsabilidad clínica demostrada, tanto en casos de HAPF como en pacientes con HAPI esporádica. Uno de ellos es el polimorfismo S775N14 detectado en un caso de HAPI esporádica y en otro de HAPF. A pesar de que este polimorfismo da lugar a un cambio de aminoácido, no es responsable de la enfermedad y se encuentra presente en el 3,4% de la población general (50 controles estudiados).
No se ha identificado ninguna mutación en los 3 sujetos sanos estudiados o familiares de pacientes diagnosticados de HAP fallecidos.
Caracterización fenotípica de familias con hipertensión arterial pulmonar familiarSe estudió a la familia del caso 7. El caso índice, portador de la mutación R491Q, fue diagnosticado de HAP a los 14 años. En su historia familiar se registraban una prima diagnosticada de HAP que falleció a los 19 años y 4 parientes cercanos fallecidos de muerte súbita o patología cardiaca no especificada, incluyendo la abuela materna (fig. 2). Se efectuó el estudio familiar del gen BMPR2, incluyendo un total de 28 casos de 3 generaciones, a partir de un hermano de la abuela materna (II generación) (fig. 2). En este último, se detectó la presencia de la mutación a los 65 años, presentando en ese momento un ecocardiograma normal. Posteriormente, a los 9 años de seguimiento, se detectó un incremento anómalo de la PAP en el ecocardiograma, confirmándose posteriormente el diagnóstico de HAP mediante estudio hemodinámico.
.")
En la III generación se estudiaron 15 miembros, de los cuales 8 eran portadores de la mutación. Dos de ellos también fueron diagnosticados de HAP mediante estudio hemodinámico al cabo de 5 y 7 años, respectivamente. En la última generación analizada (IV) se detectó la mutación en 5 casos de los 12 estudiados, estableciéndose el diagnóstico de HAP en el caso índice (ya descrito) y, en otro miembro, a los 3 años de edad. La edad media de presentación de la enfermedad en la II generación fue de 75 años, de 49 años en la III y de 9 años en la IV, confirmándose el fenómeno de anticipación genética.
En total, se detectó la mutación R491Q en 14 de los 28 miembros de la familia estudiados (50%). De entre los portadores de la mutación, en 5 casos (incluyendo el caso índice) se estableció el diagnóstico hemodinámico de HAP, lo que indica una penetrancia de la enfermedad en esta familia del 35,7%.
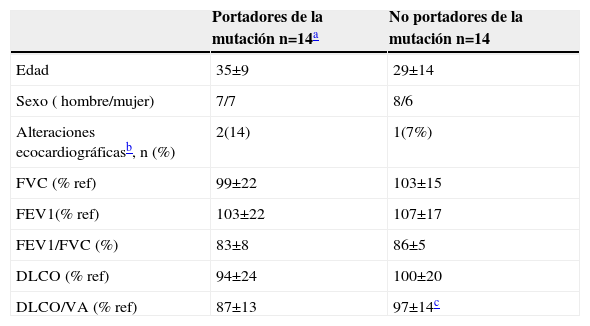
Los datos demográficos y funcionales de los sujetos de esta familia, agrupados en función de la expresión de la mutación, se detallan en la tabla 2.
Datos demográficos y funcionales de la familia con la mutación R491Q
| Portadores de la mutación n=14a | No portadores de la mutación n=14 | |
| Edad | 35±9 | 29±14 |
| Sexo ( hombre/mujer) | 7/7 | 8/6 |
| Alteraciones ecocardiográficasb, n (%) | 2(14) | 1(7%) |
| FVC (% ref) | 99±22 | 103±15 |
| FEV1(% ref) | 103±22 | 107±17 |
| FEV1/FVC (%) | 83±8 | 86±5 |
| DLCO (% ref) | 94±24 | 100±20 |
| DLCO/VA (% ref) | 87±13 | 97±14c |
DLCO: capacidad de difusión del monóxido de carbono; FEV1: volumen espiratorio forzado en el primer segundo; FVC: capacidad vital forzada; % ref: porcentaje respecto al valor de referencia; VA: volumen alveolar.
Dado el elevado número de sujetos estudiados, se investigó si en dicha familia la DLCO difería entre los sujetos asintomáticos portadores de la mutación y los no portadores. Considerados globalmente, no existían diferencias significativas entre ambos grupos. Sin embargo, un análisis de la varianza considerando como cofactor la generación, puso de manifiesto una interacción entre el valor de la relación DLCO/KCO y la generación. Por consiguiente, se compararon los valores de KCO entre los miembros de una misma generación. En los sujetos asintomáticos de la III generación se observó un valor de KCO menor en aquellos que expresaban la mutación (n=7), comparado con los que no la expresaban (n=7) (88±5% y 104±9% del valor de referencia, respectivamente; p<0,01) (fig. 3).

También se estudiaron simultáneamente 5 familiares del caso 5, sin conocimiento del estudio genético del caso índice. No se detectaron mutaciones del gen BMPR2 en dicho caso índice y, como podía anticiparse, tampoco en sus familiares.
DiscusiónEl presente estudio es el primero que analiza la prevalencia de las mutaciones del gen BMPR2 en pacientes con HAPF en población española. Se han detectado tan solo dos mutaciones en 8 casos de HAPF, lo que indica una frecuencia de mutaciones del 25% en dicho grupo. Este porcentaje es bajo si lo comparamos con otros datos publicados, que indican que hasta un 70% de los individuos con HAPF presentan mutaciones en el gen BMPR215. Una posible explicación a esta diferencia podría ser la presencia de mayor heterogeneidad genética en nuestra población, lo que implicaría la contribución de otros genes no estudiados en la patogénesis de la HAP. Otro hecho a destacar es la limitación técnica de los SSCPs, ya que el estudio mediante esta técnica ofrece un 80% de eficacia en relación con la secuenciación directa, lo que disminuiría el porcentaje de detección de mutaciones empleando la misma16. Tampoco podemos descartar la presencia de mutaciones en regiones intrónicas no estudiadas, o bien grandes deleciones o duplicaciones.
El hecho de disponer de una familia en la que ha sido posible estudiar a 28 miembros ha permitido evidenciar el fenómeno de la anticipación genética (inicio de la enfermedad a edades más jóvenes y/o mayor gravedad clínica en las generaciones subsecuentes). La mutación R491Q se identificó en 14 de los individuos analizados, lo que corresponde al 50% sobre el total de esta familia. El diagnóstico de HAP se estableció en 5 (35,7%) de los portadores estudiados, incluyendo el caso índice, lo que representa un porcentaje de penetrancia de la enfermedad algo superior a lo publicado en otros estudios15. De todos modos, en términos generales la penetrancia puede considerarse reducida, lo que indica que no todos los individuos que son portadores de la mutación van a desarrollar la enfermedad. Este hecho sugiere que la mutación del gen es requerida pero no suficiente para expresar la enfermedad17. Posiblemente, en la patogénesis de la enfermedad podrían contribuir otros factores como, por ejemplo, mediadores inflamatorios o ambientales, que actuarían como modificadores de la enfermedad ante un genotipo permisivo18,19.
En el grupo de pacientes con HAPI, la prevalencia de mutaciones del gen BMPR2 fue del 11%. Este hallazgo concuerda con estudios previos, aunque los estudios publicados muestran resultados dispares, con prevalencias que oscilan entre el 11% y el 40%7,13. En un primer estudio, Thomson y et al6 observaron mutaciones del gen BMPR2 en 13 pacientes de 50 diagnosticados de HAPI (26%). En el estudio efectuado en población española citado anteriormente10, 2 de los 8 pacientes con HAPI estudiados mostraron mutaciones, y un tercero mostró un cambio sin responsabilidad demostrada en el desarrollo de HAP. Por consiguiente, la frecuencia de mutaciones en esta otra población española de pacientes con HAPI esporádica (25%) es similar a la obtenida en nuestro estudio en casos de HAP familiar y más elevada que en los casos con HAPI esporádica. De todos modos, al tratarse de series con un número de casos muy reducido no es posible extraer conclusiones con relación a estas diferencias.
No se detectó ninguna mutación en los individuos sanos con historia familiar de HAP que nos fueron remitidos para posible consejo genético, ni en los familiares del caso 5, que fueron estudiados antes de conocer si el caso índice era portador de mutación o no. Estos datos confirman el hecho de que para efectuar un correcto cribado familiar en la HAP es preciso el diagnóstico genético de certeza del caso índice15.
Sin duda, el hallazgo de mayor interés clínico del presente estudio fue la diferencia en la capacidad de difusión de CO observada entre portadores y no portadores de la mutación. Esta es una observación novedosa, no descrita previamente. Aunque no puede afirmarse que esta alteración corresponda a una manifestación precoz de la enfermedad, sí que es concebible que la presencia de la mutación otorgue una mayor susceptibilidad a presentar anomalías funcionales en la circulación pulmonar (desventaja fisiopatológica)20, hasta ahora poco estudiadas. Estudios en modelos animales indican que la mutación del gen BMPR2 impide un adecuado remodelado del lecho vascular pulmonar ante un estímulo externo como la hipoxia prolongada21. También se ha observado un incremento significativo de la PAP durante el esfuerzo en sujetos asintomáticos portadores de la mutación, cambio que no se produjo en miembros de la misma familia no portadores de la mutación11. Sztrymf et al22 analizaron la influencia de la mutación del gen BMPR2 en el curso clínico y el pronóstico de 223 pacientes diagnosticados de HAP familiar o HAPI esporádica, comparando portadores de la mutación (n=68) con no portadores (n=155). Los portadores de la mutación presentaban una forma más grave de la enfermedad, con edad de inicio más temprana y mayor compromiso hemodinámico en el momento del diagnóstico. Asimismo, estos últimos respondían en menor proporción a la prueba vasodilatadora aguda y eran más propensos a requerir tratamiento con prostanoides endovenosos o trasplante pulmonar22.
Hasta el momento, no se dispone de ningún biomarcador o medida funcional respiratoria que permita detectar precozmente las alteraciones que ocurren en la vasculatura pulmonar en los portadores de la mutación del gen BMPR2. Se ha sugerido que la disminución de la presión parcial de CO2 al final de la espiración durante el ejercicio podría detectar de forma temprana esta alteración, al poner de manifiesto una hipoperfusión relativa en relación con la ventilación23. Los resultados de nuestro estudio son consistentes con este concepto, al observar diferencias significativas en relación con la KCO y no con la DLCO, aunque manteniéndose dentro de los límites de referencia. Son necesarios estudios en un número mayor de portadores de la mutación del gen BMPR2 para comprender mejor la significación de este hallazgo.
En resumen, el presente estudio demuestra una baja prevalencia de mutaciones del gen BMPR2 en los pacientes con HAPF de nuestro medio evaluados. En los pacientes con HAPI esporádica la prevalencia de mutaciones también es baja, aunque concuerda con la que se ha descrito en otras áreas geográficas. Asimismo, nuestro estudio pone de manifiesto la existencia de una relación entre el valor de la KCO y la presencia de mutaciones del gen BMPR2 en portadores asintomáticos, lo que sugiere una posible alteración del lecho vascular pulmonar en estos individuos. Estudios en una muestra más amplia de pacientes permitirían valorar el papel complementario de esta medición en el cribaje y seguimiento de la HAP familiar.
Los autores agradecen a Carmen Tarancón, Ana Celia Barnosi, José Poveda, Antonio Román y Carlos Disdier su valiosa aportación de casos para este estudio.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.







