







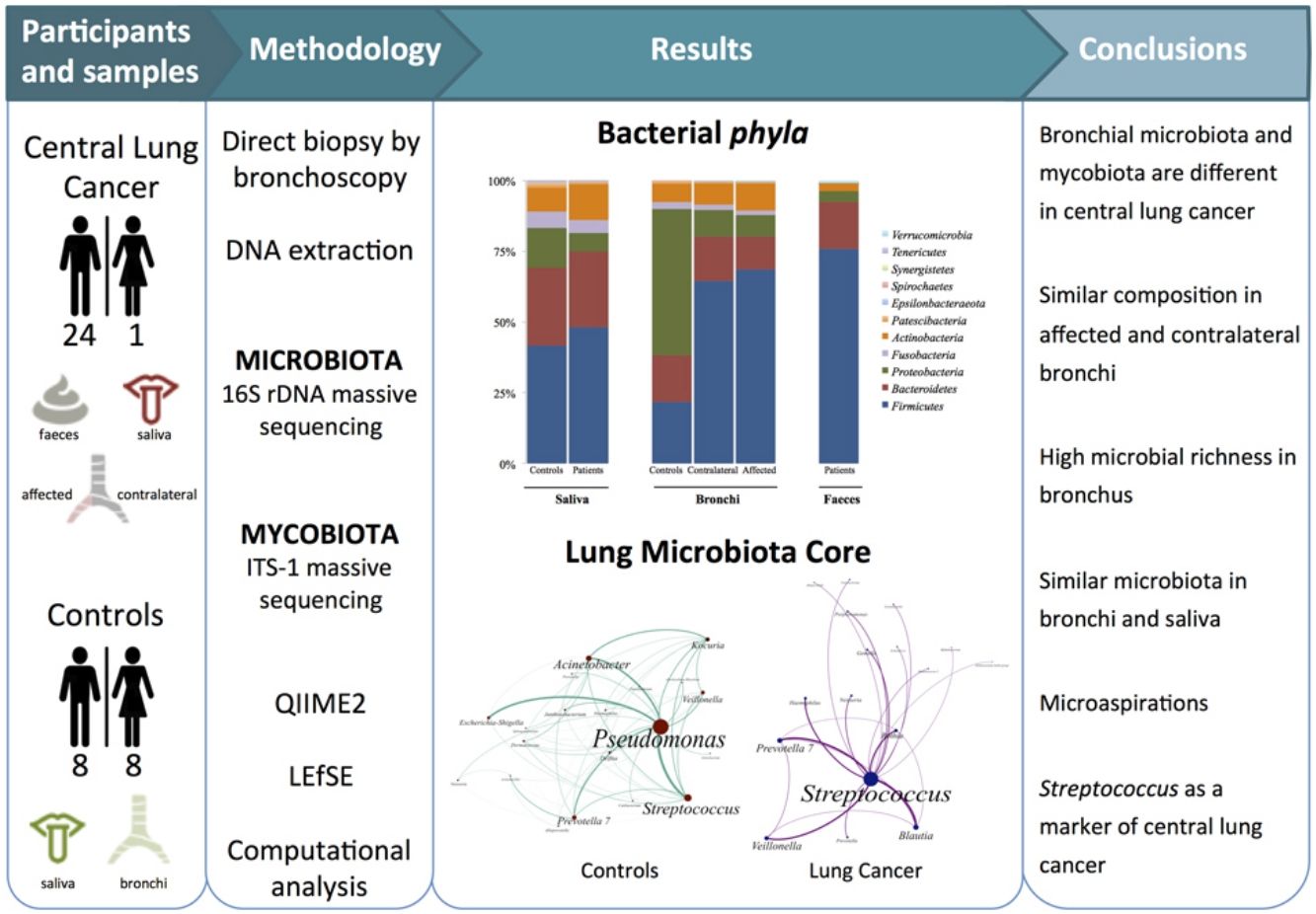
Dysbiosis in lung cancer has been underexplored. The aim of this study was to define the bacterial and fungal microbiota of the bronchi in central lung cancer and to compare it with that of the oral and intestinal compartments.
MethodsTwenty-five patients with central lung cancer and sixteen controls without antimicrobial intake during the previous month were recruited. Bacterial and fungal distribution was determined by massive sequencing of bronchial biopsies and saliva and faecal samples. Complex computational analysis was performed to define the core lung microbiota.
ResultsAffected and contralateral bronchi of patients have almost identical microbiota dominated by Streptococcus, whereas Pseudomonas was the dominant genera in controls. Oral and pulmonary ecosystems were significantly more similar in patients, probably due to microaspirations. Streptococcal abundance in the bronchi differentiated patients from controls according to a ROC curve analysis (90.9% sensitivity, 83.3% specificity, AUC=0.897). The saliva of patients characteristically showed a greater abundance of Streptococcus, Rothia, Gemella and Lactobacillus. The mycobiome of controls (Candida) was significantly different from that of patients (Malassezia). Cancer patients’ bronchial mycobiome was similar to their saliva, but different from their contralateral bronchi.
ConclusionsThe central lung cancer microbiome shows high levels of Streptococcus, and differs significantly in its composition from that of control subjects. Changes are not restricted to tumour tissue, and seem to be the consequence of microaspirations from the oral cavity. These findings could be useful in the screening and even diagnosis of this disease.
La disbiosis en cáncer pulmonar no ha sido suficientemente estudiada. Los objetivos de este estudio fueron definir la microbiota bacteriana y fúngica de bronquios con cáncer central de pulmón, y compararla con la del compartimento intestinal en heces y saliva.
MétodosSe reclutaron 25 pacientes con cáncer central de pulmón y 16 controles sin exposición antibiótica durante el mes anterior. Se determinó la composición de bacterias y hongos en biopsias de bronquio, saliva y heces. Se realizó un análisis computacional para definir el núcleo de microbiota del pulmón.
ResultadosLos bronquios afectados y contralaterales de pacientes presentaron una microbiota similar dominada por Streptococcus, mientras que Pseudomonas destacó en los controles. Los ecosistemas orales y pulmonares fueron significativamente más parecidos en pacientes, probablemente debido a microaspiraciones. La abundancia bronquial de estreptococos permitió diferenciar a los pacientes de los controles mediante una curva ROC (90,9% de sensibilidad, 83,3% de especificidad, AUC=0,897). La saliva de los pacientes presentó mayor abundancia de Streptococcus, Rothia, Gemella y Lactobacillus. El micobioma de los controles (Candida) fue significativamente diferente al de los pacientes (Malassezia), con los bronquios afectados por el cáncer similares a su saliva, pero diferentes de sus bronquios contralaterales.
ConclusionesEn el cáncer de pulmón central hay enriquecimiento de Streptococcus, y su composición es significativamente diferente de sujetos control. Las alteraciones no se limitan al tejido tumoral, y parecen ser consecuencia de microaspiraciones desde la cavidad oral. Estos hallazgos podrían ser útiles para la detección e incluso el diagnóstico de esta patología.
Culture-independent techniques have revealed the composition of a stable microbiota within the distal airways; and although normality criteria have not yet been established, atypical compositions linked to certain respiratory diseases have been detected.1 Dysbiosis is typically detected surrounding tumours tissues, and this trait has been poorly explored in the respiratory airway tissues,2,3 mostly using surgical samples.4–8 An understanding of the microbiota is necessary to decipher its possible causal role in cancer, and also to elucidate the prognosis and response to immunomodulatory therapies, because microorganisms and/or their metabolites shape the local microenvironment, influence the immune response, and impact the final battle against cancer.9 Finally, the presence and/or abundance of specific bacteria could be used as a biomarker, as occurs with Streptococcus gallolyticus sbsp. gallolyticus in the colorectal cancer.10
The aims of the present study were to define the bacterial and fungal microbiota of central lung cancer, in relation to the corresponding contralateral bronchus and compared with controls without cancer history, also defining the core of those microbiotas; and to correlate the pulmonary microbiota in lung cancer with the salivary and faecal compartments.
MethodsPatients and samplesTwenty-five patients (24 men, mean age of 68 years) diagnosed with central lung cancer, directly visible and biopsied by bronchoscopy, were recruited (Table 1). The exclusion criteria included the intake of antibiotics, pre or probiotics, and systemic corticoids during the previous 4 weeks; acute infection; radio or chemotherapy in the last year, and immunodeficiency. Tumours were histologically classified into non-small cell lung cancer (n=18, including 10 squamous, 4 adenocarcinoma, and 4 undifferentiated); and small cell lung cancer (n=7). Each patient contributed with 4 samples: (i) saliva collected from a rinse with sterile distilled water, prior to the bronchoscopic procedure, (ii) biopsies of affected, and (iii) contralateral bronchi, and finally (iv) a faecal sample (provided by 18 out of the 25 patients). The nasal route, or oral if not possible, with local instillation of lidocaine were used for bronchoscopies. Prior to the tumour sampling, biopsies for microbiota determination were obtained from contralateral non-affected tissues.
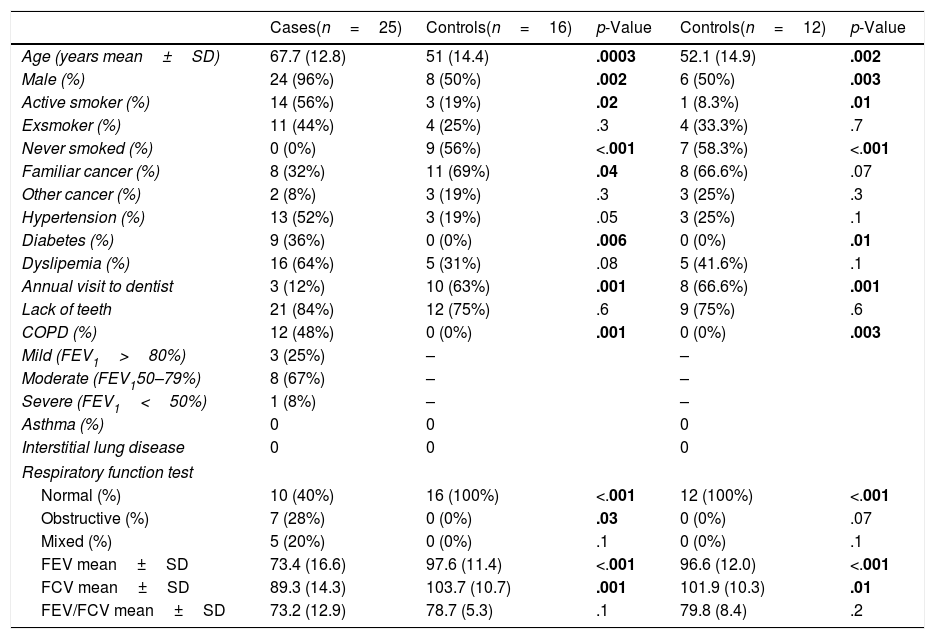
Demographic and clinical data of patients with lung cancer and healthy controls, differentiating between the entire control population (n=16) and those included in the analysis of bacteria and fungi (n=12).
| Cases(n=25) | Controls(n=16) | p-Value | Controls(n=12) | p-Value | |
|---|---|---|---|---|---|
| Age (years mean±SD) | 67.7 (12.8) | 51 (14.4) | .0003 | 52.1 (14.9) | .002 |
| Male (%) | 24 (96%) | 8 (50%) | .002 | 6 (50%) | .003 |
| Active smoker (%) | 14 (56%) | 3 (19%) | .02 | 1 (8.3%) | .01 |
| Exsmoker (%) | 11 (44%) | 4 (25%) | .3 | 4 (33.3%) | .7 |
| Never smoked (%) | 0 (0%) | 9 (56%) | <.001 | 7 (58.3%) | <.001 |
| Familiar cancer (%) | 8 (32%) | 11 (69%) | .04 | 8 (66.6%) | .07 |
| Other cancer (%) | 2 (8%) | 3 (19%) | .3 | 3 (25%) | .3 |
| Hypertension (%) | 13 (52%) | 3 (19%) | .05 | 3 (25%) | .1 |
| Diabetes (%) | 9 (36%) | 0 (0%) | .006 | 0 (0%) | .01 |
| Dyslipemia (%) | 16 (64%) | 5 (31%) | .08 | 5 (41.6%) | .1 |
| Annual visit to dentist | 3 (12%) | 10 (63%) | .001 | 8 (66.6%) | .001 |
| Lack of teeth | 21 (84%) | 12 (75%) | .6 | 9 (75%) | .6 |
| COPD (%) | 12 (48%) | 0 (0%) | .001 | 0 (0%) | .003 |
| Mild (FEV1>80%) | 3 (25%) | – | – | ||
| Moderate (FEV150–79%) | 8 (67%) | – | – | ||
| Severe (FEV1<50%) | 1 (8%) | – | – | ||
| Asthma (%) | 0 | 0 | 0 | ||
| Interstitial lung disease | 0 | 0 | 0 | ||
| Respiratory function test | |||||
| Normal (%) | 10 (40%) | 16 (100%) | <.001 | 12 (100%) | <.001 |
| Obstructive (%) | 7 (28%) | 0 (0%) | .03 | 0 (0%) | .07 |
| Mixed (%) | 5 (20%) | 0 (0%) | .1 | 0 (0%) | .1 |
| FEV mean±SD | 73.4 (16.6) | 97.6 (11.4) | <.001 | 96.6 (12.0) | <.001 |
| FCV mean±SD | 89.3 (14.3) | 103.7 (10.7) | .001 | 101.9 (10.3) | .01 |
| FEV/FCV mean±SD | 73.2 (12.9) | 78.7 (5.3) | .1 | 79.8 (8.4) | .2 |
Statistically significant values that can differentiate control cases are highlighted in bold.
Simultaneously, 16 controls without history of cancer were included, and each contributed the following: (i) oral microbiota and (ii) a single biopsy of their healthy bronchi. Controls without respiratory symptoms (except 2 with chronic cough) underwent bronchoscopy for non-cancer related indications (benign tracheal stenosis 9, fake haemoptysis 3, chronic cough 2, control of a previous endobronquial hamartoma resection 1, and dyspnoea 1), and all of them had normal spirometries. All samples were immediately frozen at −80°C after collection.
Microbiota compositionTotal DNA was obtained by the QiaAmp kit (Qiagen) from the biopsies, from the pellet of saliva after centrifugation, and from 200μl aliquots of a solution of 0.5 gr of faeces in 5ml of water. Bacterial composition was determined by PCR amplification of the 16S rDNA V3-V4 region using published primers,11 whereas the mycobiome was only analyzed in bronchial and saliva of the 16 controls and in a subset of 6 patients by amplification of the ITS-1 region.12 PCR products were submitted to massive sequencing (2× 300bp) on a MiSeq (Illumina, San Diego, CA, USA) platform, at FISABIO (Valencia, Spain). Raw sequence data were deposited in GenBank (BioProjects PRJNA586753 and PRJNA586768. QIIME2 software suite (2019.1 distribution)13 and LEfSE14 were used for analysis, and adequate negative sequencing controls were added in each process and run. A computational analysis has developed to define the microbiota core that was present in at least 95% of the individuals. This analysis is available at https://github.com/JJ-Lab/Cancer_Lung_Microbiota website.
ResultsSample filteringThree samples from affected bronchi (patients 23, 24, and 25) and four samples from control bronchi (controls 2, 3, 7, and 16) were excluded of the analysis since they did not reach minimal sequencing depth requirements (>1000reads/sample).
Alpha diversityAlpha diversity indexes, as Chao1 and Shannon, express the mean diversity in microbial species in a single community, with the highest values corresponding to the greater number of species (richness). Faeces and saliva had similar alpha diversity values, while bronchi were significantly more diverse according to Faith's PD index (which specifically assessed phylogenetic diversity) (Fig. 1). The diversity of saliva was comparable in patients and controls.
Microbiota composition
Beta diversity analysis compares the microbiota composition between different samples, and use to be referred to phyla (Fig. 2) and genera levels (Fig. 3). Patients’ saliva presented a higher density of Firmicutes and Actinobacteria to the clear detriment of Proteobacteria, and this pattern was reproduced in the affected and contralateral bronchi. Faecal samples had a remarkably high proportion of Firmicutes (>75%) (Fig. 2).


Up to 213 genera were detected in saliva samples, with 13 comprising the majoritarian in both controls and patients: Streptococcus (19% and 23%, respectively), Prevotella (15–13%), Rothia (4–7%), Veillonella (7–8%), Neisseria (6–4%), Porphyromonas (6–4%), Haemophilus (6–2%), Gemella (3–5%), Fusobacterium (5–3%), Alloprevotella (4–1%), Actinomyces (2–2%), Granulicatella (1–2%), and Leptotrichia (1–2%). The remaining genera represented less than 1% of the total microbiota abundance.
Saliva microbiota core from patients and controls were similar, and although significant differences on the bacterial proportions were detected (Figs. 3 and 4, and supplementary material), that differences are linked to relative abundance of common taxa and no to presence or absence of specific taxa. Streptococcus, Rothia, Gemella and Lactobacillus abundances are capable of distinguishing the saliva of patients from controls.
Bronchi; ROC curves of the relative Streptococcus abundance to discriminate patients from controls (B), and distance between saliva and lung microbiota in controls and in patients (C).")
More than 450 bacterial genera were identified with different distribution among patients and controls, while the microbiota of the cancer-affected bronchus was almost identical to its contralateral counterpart (Figs. 3 and 4, and Supplementary material). Particularities depending on the histological type of cancer were not detected.
Lung cancer-related genera were Streptococcus (19% in affected bronchus and 24% in contralateral), Prevotella (9–9%), Blautia (5–4%), Veillonella (4–5%), Rothia (3–4%), Neisseria (2–3%), Gemella (2–2%), and Porphyromonas (2–2%). Health bronchi from control group was dominated by Pseudomonas (21%), followed by Streptococcus (8%), Actinobacter (6%), Veillonella (4%), Prevotella (3%), Delftia (3%), Janthinobacterium (3%), Escherichia-Shigella (2%), Haemophilus (2%), and Neisseria (2%).
Streptococcal abundance characterizes the microbiota of patientsStreptococcus is a genus within the Firmicutes phylum that has a complex taxonomy. Our results showed that bronchial streptococcal abundance systematically distinguished patients from controls Up to 95 different amplicon sequence variants (ASV) assigned to Streptococcus were identified among all samples, and their particular distribution allowed us to hypothesize an exchange between oral and bronchial ecosystems (Supplementary material) Faeces were the most remote niche, with saliva and bronchi also distant. It is important to note that >95% of the ASV detected in the patients’ bronchi were also present in their saliva, while on the contrary only 36% from saliva were also in the lung, indicating that saliva is the main source of pulmonary Streptococcus. An ROC curve representing Streptococcus bronchial abundance distinguished patients from controls, particularly when a relative abundance>14.6% predicted lung cancer with 90.9% sensitivity and 83.3% specificity (AUC=0.897) (Fig. 5). Unfortunately, this result was not reproduced in saliva.

The comparison of the whole microbiota in samples within each individual revealed that saliva and bronchi of patients with lung cancer were significantly the closest ecosystems (p<0.001) (Fig. 5).
Mycobiome compositionFungal characterization through ITS1 sequencing was determined in the 16 controls (saliva and bronchus) and a subset of 6 patients (saliva, affected and contralateral bronchi). Regarding to alpha diversity, differences in Chao1 index were not observed, whereas Shannon was significantly higher in affected bronchi from patients compared to controls (p<0.03) in accordance with the results obtained for bacteria (Supplementary material).
Affected bronchi and saliva from patients have similar fungal composition, but different from both their contralateral bronchi (p<0.006), and the bronchi (p<0.001) and saliva (p<0.007) from controls. This trend supports the aforementioned bacterial findings, which also suggest an interconnection between both anatomical ecosystems in lung cancer patients. A differential abundance analysis between affected bronchi from patients and from controls showed an enrichment of Malassezia in patients, whereas controls had a higher abundance of Candida (Fig. 6).
Discussion of the bacterial microbiota between patients and controls in bronchi and saliva. Only taxa with an LDA score>4 are shown.")
The role of microbiota in carcinogenic processes has not yet been elucidated and will probably be different for each tumour and localization. Recent data clearly indicate that the microbiota contributes to the prognosis of cancer and determines the response to treatments, particularly responses to the new immunomodulation therapies.9,15 Criteria for the normal composition of lung microbiota have not yet been established, but the available data indicate that their composition in cancer patients differs considerably from that of healthy individuals.2–8 Sampling in the respiratory system requires invasive methods, and here we have characterized the respiratory microbiota surrounding central lung cancer by direct sampling of the tumour tissue by bronchoscopy. Our results reinforce the previously known particularities of the lung microbiota, which considerably differ from oral and stool microbial communities.16 Our results also allowed us to rule out significant contamination with the upper microbiota during the bronchoscopy, as other authors had previously suggested.17–19 Moreover, we considered strict inclusion criteria to avoid the possible bias of antibiotic or corticosteroid therapy, and the bacterial exchange between the anatomically separated niches such as saliva and faeces. Finally, the mycobiome composition of central lung cancer was studied.
Lung microbiota has been described based on health status or a lung cancer diagnosis in sputum,20 bronchoalveolar lavage,5 protected specimen brushing,6 cytological brushing,21 and surgical tissue.7,8,22 Our major contribution is the depiction of the microbiome surrounding central cancer via direct sampling, but our results are not necessarily applicable to the microbiota of the distal airway. Curiously, higher diversity indexes have been detected in bronchi than in faecal or oral compartments, contrarily to the biomass decreases from upper to lower tract described in healthy people.19
We found significantly higher alpha diversity values in cancer than in the control group, whereas other authors have published analogous8 and opposing results.5,6,22 Moreover, advanced cancer stages16 and reduced recurrence-free survival and disease-free survival22 have been associated with higher values of alpha diversity. In that sense, 80% of our patients were at tumour stage III or IV, and their mean survival was only of 198 days in the follow up. Furthermore, other factors such as environmental exposure, residence in high-population density areas,4,16 and pack-years of tobacco smoking, can increase the biodiversity of the lung microbiota, whereas chronic bronchitis reduces it.16 All our patients had been smokers, while most of the controls (56%) had not (Table 1).
In terms of beta diversity, cancer affected and contralateral bronchi had very similar compositions, probably reflecting the environmental influence which is not restricted to the cancer area.2,6,21,22 Although the limited size of our sample prevents us from reaching solid conclusions, no differences were detected in the composition of the bronchial microbiota as a function of the histological variants of the cancer. The abundance of Firmicutes to the clear detriment of Proteobacteria was the most noticeable result in our patients, and this result was consistent in all samples. Proteobacteria dominance in health lung microbiota, especially Pseudomonas, has been also previously corroborated.5,6,23 Higher concentrations of Streptococcus, Blautia, Akkermansia, and Rothia were observed in patients, but Streptococcus abundance was consistently the major marker linked to lung cancer. This fact has been previously reported in saliva,5 sputum,20,24 bronchoalveolar lavage,5 lung tissue,7 and protected specimen brushing.6,21 The exhaustive phylogenetic analysis of ASVs allows us to suggest that the streptococcal variants present in lung tissue are similar to those found in saliva or faeces. New studies including Streptococcus cultures and molecular characterization of the species are needed to decipher whether oral lineages are different from those found in the lungs or faeces and thus establish whether there are any markers truly associated with lung cancer, as occurs with Streptococcus gallolyticus subsp. gallolyticus and colorectal carcinoma.10,25
There is increasing evidence of a link between Streptococcus and lung cancer. Recently, Tsay et al.21 detected Streptococcus and Veillonella enrichment in the lower airways with ERK and PI3K pathways upregulation – an early event that contributes to cell proliferation, survival and tissue invasion – combining microbiome and transcriptomic signatures. The major question that a remains to be answered is whether the abundance of streptococci is a cause or consequence of the tumour process.26Streptococcus is a natural inhabitant of the oral cavity, which is connected to the lower respiratory tract by the larynx and trachea, and the oral/lung bacterial exchange could occur via microaspirations.6,21,23
Microaspiration events are common, but their frequency is significantly increased in chronic inflammatory airway diseases,23 inducing Th17 lymphocytes, as well as expression of inflammatory cytokines (as IL-1α, IL-1β and IL-17). IL-23/IL-17 axis alteration is well known in the pathogenesis of both autoimmune diseases and tumours, and Streptococcus mitis facilitate the cancer development and expansion by induction of IL-1β, IL-6, IL-10 and IL-23 transcription, Th17 activation, and an increased immune checkpoint PD-L1 expression.27 Lung resident γδ T cells with protective roles or pro-tumorigenic functions in cancer have been recently discovered, and local lung microbiota, including Streptococcus, can provoke inflammation and tumour cell proliferation via lung resident γδ T cells activation.28 Our results demonstrated a global streptococcal enrichment in patients with cancer that affected more than just the respiratory tract, supporting the idea that microorganisms can orchestrate the balance between tumour-promoted inflammation and anti-tumour immunity depending on the specific microenvironment.
Streptococcal relative abundance in bronchial biopsies was a good predictor of lung cancer, but unfortunately was not reproducible in saliva. ROC curves suggested the contralateral bronchi as the best sample (90.9% sensitivity and 83.3% specificity; AUC=0.897). Other authors found similar results in protected specimen brushing samples (87.5% of sensitivity and 55.6% specificity, AUC=0.693).6 The proportional abundance of Streptococcus should be validated in the early stages of lung cancer with subsequent follow-up to corroborate their potential use as biomarker. Along these lines, the intestinal enrichment of Streptococcus should be exhaustively explored to identify lung cancer markers in faeces.
The intestinal and the respiratory ecosystems harbour a diverse and abundant microbiota, but some particularities distinguish both ecosystems. Food intake favours a higher rate of microbial reproduction increasing the total mass that is significantly reduced after defecation. On the contrary, nutritional sources for bacteria in the airway are limited to mucus and cellular debris, while clearance is carried out by the ciliary system and the immune system, particularly by macrophages. Lung microbiota core, which is composed by those taxa that are present in >95% of individuals, had not yet been defined. We have implemented new computational strategies to define the lung microbiota core, which had not previously been defined. Our main results are the elevated alpha-diversity of the bronchial microbiota in comparison with saliva or faeces, and the dominance of Pseudomonas in controls. Presence of this particular genus is linked to cystic fibrosis, being the major pathogen that decreases the respiratory functionality within a pathogenic colonization in those patients. However, the lack of respiratory symptoms reduces the potential pathogenic role of Pseudomonas in healthy individuals, although more studies are needed in that line.
The mycobiome results were consistent with those obtained for bacteria. The fungal community was slightly richer and more diverse in patients than in controls, although the contralateral bronchus was more similar to controls than to the affected counterpart. The mycobiome of saliva and the affected bronchus from patients matched perfectly, but differed in controls, again suggesting that in patients with cancer the bronchial microbiota is the result of a continuous exchange with that of the oral niche. In terms of taxonomy, affected bronchi from patients had an enrichment of the Basidiomycota phylum with higher populations of Malassezia genus, whereas the enriched taxon in healthy individuals was the Ascomycota phylum and the genera Candida and Saccharomyces, as previously described.29 Although the public databases are increasing exponentially, it is important to note that a major limitation to describing the mycobiome is the lack of available taxonomic records. As far as we know, this is the first description of the lung mycobiome.
The main limitation of our work is that we cannot estimate the contribution effect of lung cancer factors on the bronchial microbiota, mainly tobacco (all patients had been smokers but only the half of the controls were), and COPD (12/25 patients). However, it has not been established yet if tobacco has a significant influence on lung microbiota composition, and some important studies have shown contradictory results.16,30,31 Whereas severe COPD has been linked with significant alterations in the lung microbiota composition,32,33 mild and moderate COPD (92% of our COPD patients) has been associated with Streptococcus enrichment.33 This finding might explain the association of mild/moderate COPD and lung cancer,34 although further studies are needed to confirm this association.
Additional limitations are the small number of patients, all of them from the same hospital, and the lack of other -omic analyses based on genetic expression. On the other hand, our strengths include performing the first study of microbiota combined with mycobiome of bronchial tissue obtained directly from tumour and contralateral bronchi (not adjacent to a resected tumour), as well as performing analysis of the connected ecosystems including saliva and faeces.
In summary, patients with central lung cancer have a significantly different bronchial microbiota from controls, not restricted to tumour-involved tissue, and probably conditioned by continuous microaspiration events from the oral cavity, more than by the carcinogenic process. Lung cancer was associated with a considerable enrichment of Streptococcus and we propose that this feature could be used for the screening, diagnosis of this pathology. Lung mycobiota differ considerably in control individuals, and there are dissimilarities in patients between the affected and the contralateral bronchi. An innovative bioinformatics strategy used in this study has allowed us to define healthy individuals’ the bronchial core microbiota, which is dominated by a non-pathogenic colonization of Pseudomonas.
Ethics approvalThe Ethics Committee “CEIC Aragón” approved this project in 2016 with the reference 15/2016, and all participants signed the informed consent after receiving all the information from the clinical researchers.
Authors’ contributionsSB conceived the study. JJV, ALF, EM, and AR contributed to the patients’ selection and sample collection. JMP, JG carried out the computational analysis for core microbiota definition. MPA and RdC determined the microbiota composition. AR, PG, JMP, JG, and SB participated in the analysis and interpretation of data. SB, RdC, and MPA wrote and reviewed the manuscript, and finally all authors read and approved the final version.
FundingThis work was partially supported by the project PI17/00115 which recipient is RdC. MPA was supported by the Programa Operativo de Empleo Juvenil, co-financed by the European Social Fund Investing in your future (ESF) and ERDF (PEJD-2018-PRE/BMD-8237), and by a Rio Hortega contract (CM19/00069) from the Instituto de Salud Carlos III.
Conflict of interestRdC is the recipient of a Vertex grant IIS-2017-106179 for cystic fibrosis research. The remaining authors declare that they have no competing interest.
The authors thank to Pilar García, Asunción Albericio, and the rest of nurse team of Interventional Pulmonology Unit, and to Yolanda Palacios, from Department of Microbiology of Miguel Servet Hospital for their contributions in the collection and conservation of the samples. The labour of Marta Cobo during sample processing is also recognized.
The following are the supplementary data to this article:







