

La hipertensión pulmonar (HP) es una enfermedad grave tributaria de trasplante pulmonar (TP) en su fase terminal.
Pacientes y métodosSe ha realizado un estudio retrospectivo de 15 pacientes con HP, a los que se trató con un TP en el período 1994–2004. Se revisan los datos clínicos antes del trasplante y el seguimiento tras éste.
ResultadosEn 8 pacientes (53%) la HP fue idiopática y en 2 estuvo relacionada con el consumo de aceite tóxico; el resto de etiologías, con un paciente cada una, fueron: embolia pulmonar crónica periférica, histiocitosis X, enfermedad venooclusiva, esclerodermia y cardiopatía congénita simple corregida. Los valores hemodinámicos medios fueron: presión arterial pulmonar sistólica, diastólica y media, 100; 50, y 67mmHg, respectivamente; gasto cardíaco, 2,63l/min; resistencia pulmonar total, 20,9 UW. El tiempo desde el diagnóstico de HP hasta el TP fue de 5,9 (rango: 0,4-20) años. Siete pacientes estaban en clase funcional III y 8 en clase funcional IV. La distancia media recorrida en la prueba de la marcha de 6min fue de 204m (rango: 0–360). La mortalidad perioperatoria fue de 4 pacientes (26%). La supervivencia a 1; 3, y 5 años fue de 9 (60%), 7 (46%) y 6 (40%) pacientes, respectivamente.
ConclusionesEl TP bilateral es una opción terapéutica que, en casos seleccionados, presenta resultados comparables al tratamiento médico más activo de la HP.
Pulmonary hypertension is a serious disease that, in its terminal phase, requires lung transplantation.
Patients and methodsA retrospective study was undertaken of 15 patients with pulmonary hypertension who underwent lung transplantation between 1994 and 2004. Clinical data recorded before the procedure and during follow-up were reviewed.
ResultsPulmonary hypertension was reported as idiopathic in 8 patients (53%) and related to consumption of toxic oil in 2. The remaining causes were documented as chronic peripheral pulmonary embolism, histiocytosis X, venoocclusive disease, scleroderma, and simple corrected congenital heart defect in 1 patient each. The mean values of the hemodynamic variables were 100, 50, and 67mm Hg for systolic, diastolic, and mean pulmonary artery pressure, respectively; 2.63L/min for cardiac output; and 20.9 Wood units for total pulmonary resistance. The mean time between diagnosis of pulmonary hypertension and lung transplantation was 5.9 years (range, 0.4–20 y). Seven patients were in functional class III and 8 in functional class IV. The mean 6-minute walk distance was 204m (range, 0–360m). Four patients (26%) died during the during the perioperative period and 9 (60%), 7 (46%), and 6 (40%) were still alive at 1, 3, and 5 years, respectively.
ConclusionsDouble-lung transplantation is a therapeutic option that, in certain cases, has similar outcomes to those achieved with the most aggressive medical treatment for pulmonary hypertension.
La hipertensión pulmonar (HP) es una enfermedad infrecuente y grave para la que aún no existe tratamiento curativo. Se define por la presencia de una presión de arteria pulmonar media superior a 25mmHg en reposo o de más de 30mmHg durante el ejercicio, y su evolución natural provoca el fallecimiento en una media de 3 años desde el momento del diagnóstico1,2.
Su tratamiento médico específico se ha ido desarrollando durante los últimos 30 años, pero ha sido en la presente década cuando se ha comenzado a aplicar la mayoría de los fármacos útiles para esta enfermedad. Por ello desde mediados de los años ochenta y hasta entrada la década actual la única posibilidad de tratamiento para estos pacientes era el trasplante pulmonar (TP). La modalidad de TP indicada para la HP ha variado durante estos años: en una fase inicial se indicaba mayoritariamente el trasplante cardiopulmonar, que en algunas series presenta resultados favorables3; luego algunos grupos se decantaron por el unipulmonar4, y en los últimos años es el bipulmonar el que se ha asentado como técnica de elección en la mayoría de los centros5,6,7.
En cuanto al tratamiento médico, el primer fármaco específico disponible para la enfermedad fue el epoprostenol, aprobado por las agencias reguladoras a finales de los años noventa8. A partir de 2002 aparecieron los ensayos fundamentales sobre el treprostinil subcutáneo9, el iloprost inhalado10, el beraprost oral11 y el bosentán12. Desde 2005 se ha publicado ensayos clínicos con sildenafilo13, sitaxentán14 y ambrisentán15. Estos fármacos han demostrado mejorar parcialmente la capacidad de ejercicio y los parámetros hemodinámicos, pero no todos los tratados obtienen un beneficio sostenido, lo que evidencia que estamos lejos de alcanzar la curación de la enfermedad. Por todo esto, el TP sigue vigente como opción final en el esquema terapéutico de la HP16. Un problema grave que ha surgido con la aparición de los nuevos tratamientos y sus combinaciones es cuándo evaluar a un paciente para el eventual trasplante. Todos los grupos de trasplante están de acuerdo en que el paciente no debe llegar al TP en situación terminal, pues esto imposibilita su realización17. Aunque el TP no está exento de mortalidad, en estos pacientes consigue una rápida normalización de la hemodinámica del territorio pulmonar, gracias a lo cual el ventrículo derecho es capaz de recuperar su función y estructura originales18,19.
En el presente estudio se revisa la serie de pacientes afectados de HP y trasplantados en nuestro centro durante un período de 10 años.
Pacientes y métodosSe recogieron retrospectivamente los datos de los pacientes a quienes se realizó un TP por HP en nuestro centro desde agosto de 1994 a diciembre de 2004. Desde esa fecha hasta el cierre del estudio, en diciembre de 2007, no se llevó a cabo ningún TP por HP en nuestro centro. De un total de 243 trasplantes efectuados en dicho período, 15 (6%) correspondían a pacientes afectados de HP, de los que 11 (73%) eran mujeres y 4 varones (27%), con una edad media de 37,3 años (rango: 23–56) en el momento del TP. A todos se les había diagnosticado la HP mediante cateterismo cardíaco. Por etiología, y siguiendo la clasificación clínica20, 13 pertenecían al grupo I (8 casos de hipertensión pulmonar idiopática, 2 de ingesta de tóxicos —aceite de colza—, uno secundario a esclerodermia, uno de enfermedad venooclusiva pulmonar, uno de ductus arterioso corregido), uno pertenecía al grupo IV (tromboembolia pulmonar por síndrome de Paget-Schrötter) y otro al grupo V (histiocitosis X). Los valores medios de la hemodinámica pulmonar diagnóstica fueron: presión arterial pulmonar sistólica de 100 mmHg (rango: 65–141), diastólica de 50mmHg (rango: 28–72) y media de 67,3mmHg (rango: 42–97); gasto cardíaco de 2,63l/min (rango: 1,9–3,24); resistencia pulmonar total de 20,9UW (rango: 12,8–30,4). La prueba vasodilatadora aguda con epoprostenol intravenoso se realizó en 13 pacientes y fue negativa en todos ellos (tabla 1).
Tabla 1. Características de la muestra antes del trasplante
| Edad/sexo | Etiología | Fecha del trasplante | Supervivencia tras trasplante | PAPm (mmHg) | GC (l/min) | CF | PM6m (m) | Tratamiento | FVC (%) | |
| 1 | 32/M | Aceite de colza | 1994 | 4.927 * | 75 | 2,2 | III | 47 | – | 86 |
| 2 | 33/M | HAPI | 1996 | 124 | 64 | 2,6 | IV | 180 | – | 85 |
| 3 | 34/V | HAPI | 1996 | 770 | 64 | 1,9 | IV | 252 | – | 107 |
| 4 | 27/M | TEP | 1996 | 1 | 49 | 2,6 | III | 323 | – | 98 |
| 5 | 23/V | HAPI | 1996 | 4.197 * | 70 | 3,2 | IV | – | Iloprost inhalado | 104 |
| 6 | 46/M | Aceite de colza | 1998 | 3.325 * | 45 | 2,9 | III | 240 | – | 103 |
| 7 | 52/M | HAPI | 1999 | 1.080 | 64 | 2,6 | IV | 228 | Epoprostenol | 110 |
| 8 | 45/M | HAPI | 1999 | 6 | 62 | 2,6 | III | 204 | – | 78 |
| 9 | 36/V | HAPI | 1999 | 37 | 64 | 3,1 | III | 210 | – | 97 |
| 10 | 26/M | Ductus arterioso corregido | 2000 | 1 | 97 | 2,4 | III | 360 | – | 62 |
| 11 | 51/M | Esclerodermia-CREST | 2000 | 2.628 * | 42 | 2,6 | III | 255 | – | 71 |
| 12 | 56/M | HAPI | 2000 | 2.615 * | 85 | 2,9 | IV | 336 | Epoprostenol | 91 |
| 13 | 34/V | EVOP | 2001 | 1.365 | 64 | 2,6 | IV | – | – | 85 |
| 14 | 39/M | Histiocitosis X | 2002 | 2.035 * | 70 | 2,6 | IV | 312 | Epoprostenol | 56 |
| 15 | 26/M | HAPI | 2004 | 1 | 78 | 2,6 | IV | 372 | Epoprostenol | 50 |
CF: clase funcional; CREST: síndrome de calcinosis cutánea, fenómeno de Raynaud, disfunción esofágica, esclerodactilia y telangiectasias; EVOP: enfermedad venooclusiva pulmonar; FVC: capacidad vital forzada; GC: gasto cardíaco; HAPI: hipertensión pulmonar idiopática; M: Mujer; PAPd: presión arterial pulmonar diastólica; PAPm: presión arterial pulmonar media; PAPs: presión arterial pulmonar sistólica; PM6m: prueba de la marcha de 6min; TEP: tromboembolia pulmonar; V: varón.
* Pacientes vivos al cierre del estudio.
Estos pacientes se consideraron candidatos al TP por cumplir los criterios establecidos, ya que presentaban un estado avanzado de la enfermedad con escasa esperanza de vida, tenían menos de 60 años, no presentaban enfermedad progresiva e irreversible en otros órganos, infecciones activas ni contraindicaciones para el tratamiento inmunodepresor, tenían un adecuado estado nutricional y no se evidenciaron contraindicaciones quirúrgicas ni problemas psicosociales. Se les estudió mediante el protocolo de evaluación preoperatoria del hospital y se les incluyó en lista de espera de TP, previa aceptación del Comité de TP de nuestro centro.
Para esta indicación sólo se consideró la posibilidad de trasplante bipulmonar, cuya técnica se ha descrito con anterioridad21. Brevemente, se utiliza un acceso por toracoesternotomía anterior transversal y submamaria (clamp shell) e implante secuencial. En todas las intervenciones se indicó circulación extracorpórea electiva.
En todos los casos el tratamiento inmunodepresor postoperatorio consistió en la combinación de ciclosporina, metilprednisolona y azatioprina, mientras que el tacrolimus, micofenolato de mofetilo y rapamicina se reservaron para la segunda línea de inmunodepresión. Los fármacos utilizados durante la fase de inducción fueron los mismos que los administrados en la fase de mantenimiento. El tratamiento con ciclosporina se inició el día del trasplante ajustando la dosis a un valor residual (C0) entre 200 y 400ng/ml. El tratamiento con metilprednisolona se inició durante la intervención, antes de la reperfusión del injerto, a dosis de 10 mg/kg; la dosis del primer día fue de 375mg, la de los primeros 3 meses de 0,5mg/kg/día, y con posterioridad se administraron de forma indefinida 0,1–0,2mg/kg/día. Todos los pacientes intervenidos antes del año 2000 recibieron profilaxis anticitomegalovirus con ganciclovir intravenoso; entre 2000 y 2002 recibieron ganciclovir oral, y entre 2002 y 2004 se empleó valganciclovir durante un período de 4 a 6 meses tras la intervención.
Se ofrece una descripción retrospectiva de los resultados de este grupo de pacientes mediante la revisión sistemática de sus historias clínicas. Para el estudio descriptivo las variables cuantitativas se presentan como media y desviación estándar, indicándose el rango y/o la mediana cuando pueden aportar información. Las variables cualitativas se expresan como frecuencia y porcentaje. Para los cálculos de la supervivencia se utilizó el método de Kaplan-Meier.
ResultadosDesde agosto de 1994 a febrero de 2004 se realizaron 15 TP por HP en nuestro centro, lo que supone un 6% del total de TP practicar en dicho período. El tiempo desde el diagnóstico de HP hasta el TP fue de 5,9 (rango: 0,4-20) años. El tiempo medio en lista de espera fue de 222 días (rango: 6–684) días. En el momento del trasplante 7 pacientes se encontraban en clase funcional III y 8 en clase IV. Cinco pacientes, todos ellos en clase funcional IV, recibían tratamiento específico para la HP con prostaglandinas (en 4 casos por vía intravenosa y en uno inhalada). Los 13 pacientes aptos para realizar la prueba de la marcha de 6min recorrieron una distancia media de 255m (rango: 47–372). Dos pacientes no pudieron realizarla por encontrarse en situación crítica. En el momento de su inclusión en lista de espera de TP los pacientes presentaban una capacidad vital forzada media del 85% (rango: 50–110%), volumen espiratorio forzado en el primer segundo del 83% (rango: 49–112%) y capacidad de difusión del monóxido de carbono del 58% (rango: 21–100%).
En todos los casos se indicó trasplante bipulmonar con circulación extracorpórea electiva. El tiempo medio en circulación extracorpórea fue de 386min (rango: 180–480) y el de isquemia del segundo pulmón de 500min (rango: 290–570). No hubo mortalidad intraoperatoria. Tras la intervención se aplicó ventilación mecánica durante una media de 270h (rango: 8–1.680). De los pacientes que superaron el postoperatorio inmediato, se extubó a 6 (54,5%) en los 7 días posteriores a la intervención.
La estancia hospitalaria media tras el TP fue de 38 días (rango: 1–150), repartidos entre 22 días (rango: 1–112) en la Unidad de Cuidados Intensivos y 16 (rango: 0–68) en planta de hospitalización. Si valoramos el grupo de pacientes que finalmente recibieron el alta hospitalaria, la estancia media global fue de 46 días (rango: 15–150), repartidos entre 19 días (rango: 5–82) en la Unidad de Cuidados Intensivos y 26 (rango: 8–43) en planta de hospitalización.
Complicaciones perioperatoriasLa mortalidad perioperatoria fue de 4 pacientes (26,6%), de los que 3 fallecieron en las 24h posteriores a la intervención. Las causas fueron hemorragia pleural masiva posquirúrgica (n=2), tromboembolia pulmonar (n=1) y disfunción primaria del injerto (n=1).
El 86% de los pacientes presentó alguna complicación en el período perioperatorio. En 7 pacientes (46%) se observaron complicaciones pleuropulmonares (hemorragia pleural, dehiscencia de sutura, hemorragia de sutura, atelectasia, isquemia de sutura). En 4 pacientes (26%) se evidenciaron complicaciones hemodinámicas (lesión de preservación grave, tromboembolia pulmonar, taponamiento pericárdico), y 3 (20%) presentaron complicaciones hematológicas (plaquetopenia persistente, coagulopatía de consumo, diátesis hemorrágica postoperatoria).
Durante este período 5 pacientes (33%) presentaron complicaciones infecciosas (2 casos de bacteriemia por Staphylococcus aureus, uno de bronquitis por Candida, uno de bronquitis por Pseudomonas aeruginosa, uno de bronquitis por varios microoorganismos (Stenotrophomona maltophilia, Acinetobacter spp. y Candida spp.), y hubo una sepsis por Acinetobacter spp.
Complicaciones tardíasLa mortalidad tardía (desde el día 30 postrasplante hasta el cierre del estudio) fue de 5 pacientes, de los que 3 fallecieron por bronquiolitis obliterante (BOS), uno por toxoplasmosis el día 37 del postoperatorio y otro por insuficiencia multiorgánica el día 124. Tras el primer mes de seguimiento, 7 de 11 pacientes presentaron en total 23 episodios de infección, con una media de 2,09 episodios por paciente. Por etiología, hubo 15 infecciones bacterianas, 3 por Aspergillus spp., una por Candida spp., 2 por herpes zóster y 2 por citomegalovirus.
Rechazo agudo y crónicoSe diagnosticaron mediante biopsia transbronquial 5 episodios de rechazo agudo sintomático durante el primer mes, con una media de 12,6 días postrasplante (rango: 9–16). En ningún caso se observaron recidivas de rechazo agudo. La incidencia global de rechazo agudo fue del 45% (5 pacientes de los 11 que sobrevivieron más de una semana).
Se diagnosticó de BOS a 7 de los 11 supervivientes a largo plazo. La media de tiempo desde el TP hasta el diagnóstico de BOS fue de 2,4 años (rango: 0,3–5,8). La mortalidad por BOS fue de 3 de los 7 pacientes al final del estudio. Al cierre de éste, el tiempo medio de seguimiento de los pacientes diagnosticados de BOS era de 3,3 años (rango: 1,3-6,7).
SupervivenciaLa media de supervivencia fue de 4,2 años (figura 1). Cuatro pacientes fallecieron durante el postoperatorio inmediato y 2 posteriormente durante el ingreso. De los 9 pacientes a quienes se dio el alta tras el implante, todos se incorporaron a un ritmo de vida normal e independiente, con desaparición de los parámetros de HP y de sufrimiento de las cavidades cardíacas derechas en el seguimiento a largo plazo. La media de supervivencia en este grupo fue de 6,9 años (rango: 2,1–13,5).
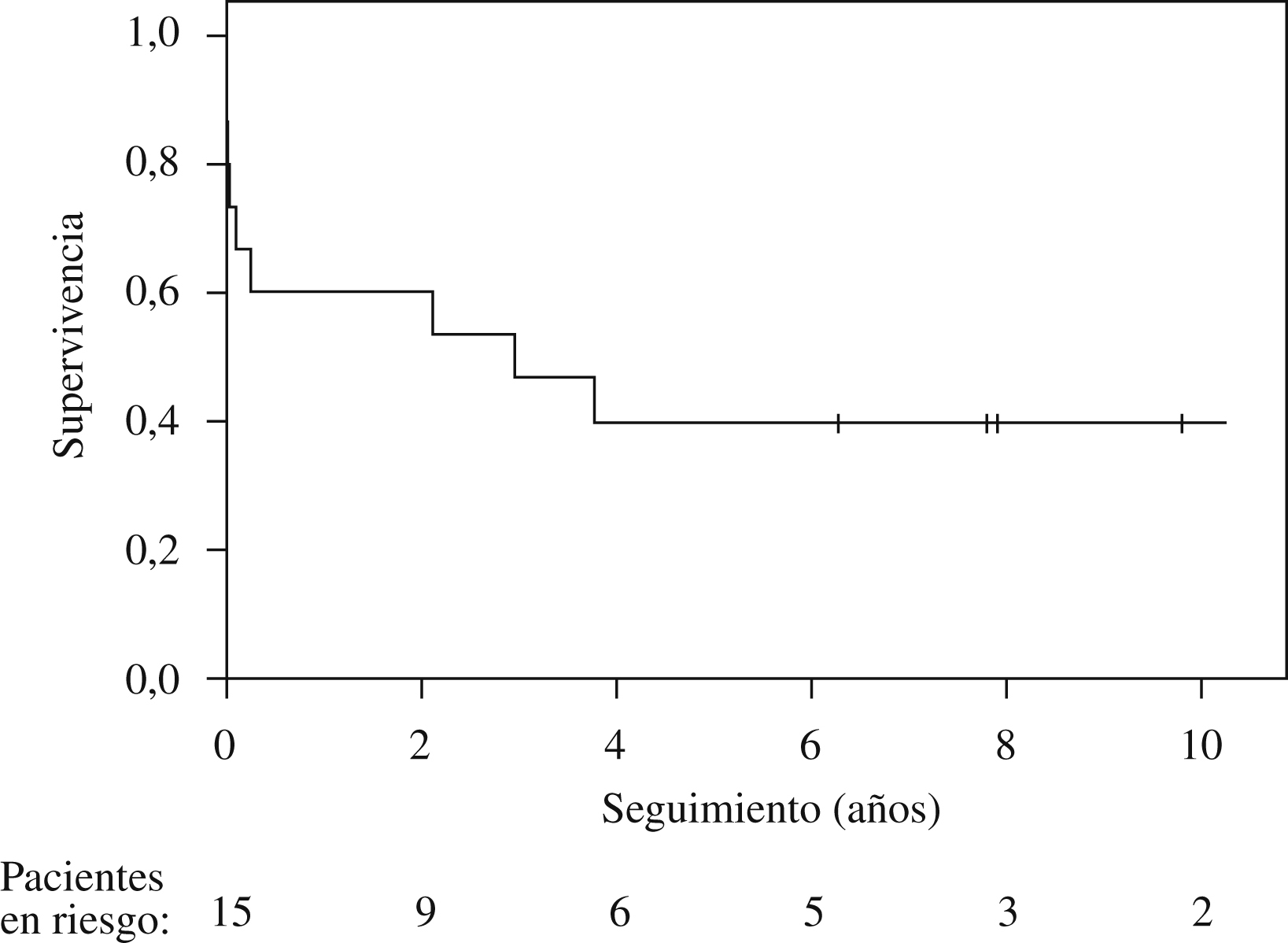
Figura 1. Supervivencia actuarial de 15 pacientes con hipertensión pulmonar a quienes se realizó un trasplante pulmonar.
En el momento del cierre del estudio, 5 pacientes (33,3%) acuden a control en nuestro centro y uno ha abandonado el seguimiento. La media de supervivencia de este grupo al cierre del estudio es de 10,8 años (rango: 5,5–13,5).
DiscusiónLa media de supervivencia a largo plazo en la presente serie es de 4,2 años. Los TP por HP representan el 6% del total de TP realizados en nuestro centro en el período descrito. Estos datos de actividad y resultados están en consonancia con los provenientes del Registro Internacional6, donde el porcentaje de trasplantes realizados por HP va desde el 10% del total de trasplante bipulmonares en los años noventa al actual 3% del total, con una supervivencia a 1 y 5 años del 65 y el 46%, respectivamente. Se observan resultados similares en otras series de pacientes: el grupo de St. Louis4 presenta —en el que puede considerarse el mayor grupo de pacientes con TP por HP en un solo centro— una serie de 100 pacientes con una supervivencia actuarial a 5 años del 57%. Estos datos contrastan claramente con los presentados por el programa de Baltimore, en los que en un reducido grupo de pacientes la supervivencia actuarial es del 80% a los 4 años22. Sea como fuere, los resultados de la presente serie están en la línea de los del Registro Internacional, que representa la mayor parte de la actividad de TP en el ámbito mundial, tanto por lo que se refiere a la supervivencia a largo plazo como a la mortalidad perioperatoria. Ésta ha sido del 26%, cifra que puede considerarse comparable a los datos publicados6. Se sabe que estos pacientes presentan un riesgo operatorio mayor. Además, en la actividad realizada durante los años noventa influye la curva de aprendizaje, cuyo efecto también se ha evaluado en el Registro Internacional, de modo que se observan mejores resultados a medida los equipos adquieren experiencia.
En cuanto a las causas de muerte en el período perioperatorio, hubo 2 hemorragias pleurales, una embolia pulmonar y una insuficiencia primaria del injerto. Salvo la embolia pulmonar, que es una complicación infrecuente, estas causas son las esperables por la enfermedad y las dificultades de su intervención. Tanto en los datos del Registro Internacional6 como en series provenientes de un solo centro4,22 la causa principal de fallecimiento, que hace que la supervivencia perioperatoria para la HP tras el trasplante sea inferior, es la disfunción primaria del injerto6,23, que en la presente serie fue causa de muerte en un solo paciente.
Las otras complicaciones perioperatorias no mortales fueron: 4 episodios de hemorragia pleural, 2 de isquemia de sutura, 2 de dehiscencia de sutura y uno de hemorragia de sutura. En este período la incidencia y las características de las complicaciones infecciosas no difirieron de las habituales para el resto de la población con TP. Respecto a las complicaciones a largo plazo, la BOS por rechazo crónico se ha descrito como más prevalente en los pacientes que reciben un TP por HP24,25, hecho concordante con nuestros datos, ya que el 63% de los pacientes con supervivencia a largo plazo presentaron BOS. Estos resultados difieren de los presentados en series de otros centros de referencia4,22: en la serie del Hospital de St. Louis la prevalencia de BOS en un grupo de 51 trasplantes bipulmonares por HP fue del 38%, y en la del grupo de Baltimore de sólo un 18% en un grupo de 55 pacientes. Estas cifras discordantes podrían explicarse por el tiempo de seguimiento, que en estas series osciló entre 3 y 5 años, mientras que la media de seguimiento de nuestros pacientes fue de 6,9 años.
La ventaja del trasplante bipulmonar frente a otras técnicas está reconocida en la literatura médica, incluso por grupos que inicialmente optaron por el TP unipulmonar4,26. El principal motivo es que el manejo postoperatorio resulta más sencillo, al observarse una menor lesión de reperfusión que en el trasplante unipulmonar. Por otra parte, algunos autores opinan que con el TP bilateral existe una mayor reserva pulmonar cuando aparece la BOS22,27. A largo plazo, parece que la ventaja más importante del trasplante bipulmonar sería una supervivencia mayor a la del unipulmonar4,6. Estos argumentos se sopesaron ya a principios de los años noventa al realizar la indicación del tipo de TP en la presente serie.
Respecto al trasplante cardiopulmonar, sus indicaciones se han visto reducidas en los últimos años, al demostrarse que la función y la estructura del ventrículo derecho se recuperan una vez corregida la hemodinámica pulmonar tras el TP18,19, por lo que actualmente su indicación queda limitada a los casos de HP asociada a síndrome de Eisenmenger causado por cardiopatía congénita compleja6,28.
Con la aparición de los nuevos tratamientos específicos para la HP se ha replanteado el papel del TP en el tratamiento de esta enfermedad. Aunque la proporción de TP indicados por HP en relación con otras enfermedades ha disminuido claramente en los últimos años, su número absoluto se mantiene estable, lo que confirma la vigencia del TP en el esquema terapéutico de la HP. Resulta difícil decidir en qué momento se debe indicar el trasplante. Es al iniciar el tratamiento con epoprostenol intravenoso cuando se plantea la posibilidad del TP, ya que la supervivencia con este tratamiento es globalmente similar a la del TP para esta enfermedad29. El TP estará indicado cuando el paciente continúe en clase funcional III o IV a pesar de recibir tratamiento con epoprostenol intravenoso16. Puesto que el tiempo en lista de espera para el TP es generalmente elevado, la evaluación del paciente para el trasplante y su inclusión en lista de espera se deben anticipar al fracaso del tratamiento intravenoso, definido por los marcadores de mal pronóstico (prueba de la marcha de 6min<380m tras 3 meses de tratamiento con epoprostenol intravenoso, aparición de derrame pericárdico, presión elevada en aurícula derecha, índice cardíaco bajo). Por todo ello, parece adecuado llevar a cabo una evaluación clínica y hemodinámica cuidadosa en los pacientes tras 3 o 4 meses de tratamiento intravenoso, lo que ayudará a establecer la indicación del TP en el momento oportuno. Si con el tratamiento con epoprostenol el paciente presenta una respuesta muy favorable, hasta alcanzar la estabilidad y recuperar una clase funcional I–II, podrá posponerse el TP. Si no es así, se debe proceder a su inclusión en lista de espera, ya que la decisión de aguardar más tiempo suele desembocar en la necesidad de un trasplante urgente, lo que empeora sensiblemente el pronóstico30.
En conclusión, los resultados del TP para la HP en nuestro centro son equiparables a los publicados hasta ahora, y estamos en condiciones de afirmar que es una opción terapéutica que, en pacientes seleccionados, ofrece resultados alentadores para una enfermedad grave y progresiva. El impacto favorable que los nuevos tratamientos han tenido en el manejo de la HP ha condicionado un descenso de la indicación del TP en los últimos años. Sin embargo, todo parece indicar que, ante la ausencia de un tratamiento médico curativo, el TP mantendrá su papel en este campo. Los profesionales centrados en esta área debemos ser sensibles a esta realidad y hacer que nuestros pacientes lleguen al TP con posibilidades reales de superarlo.







