




Introducción
La enfermedad pulmonar obstructiva crónica (EPOC) es un problema de salud pública de primera magnitud. Estudios realizados en España señalan que un 9% de la población general de entre 40 y 70 años de edad está afectada de EPOC1. Esto indica que más de un millón de personas de este grupo de edad padecen la enfermedad en España, cuya expresión patológica es, en la mayoría de los casos, el enfisema pulmonar.
La EPOC se caracteriza por una lenta y progresiva obstrucción al flujo aéreo debida a la pérdida de elasticidad resultante de la destrucción del parénquima pulmonar y la obstrucción de las vías aéreas periféricas. El humo del tabaco es el factor causal en más del 90% de los casos en los países occidentales2. A pesar de la clara asociación entre el tabaquismo y la obstrucción al flujo aéreo, existe una notable variación interindividual en la respuesta al humo del tabaco3,4. Esto indica que hay cofactores adicionales genéticos o ambientales que pueden contribuir al desarrollo de la EPOC.
Numerosos datos de estudios clínicos y epidemiológicos apuntan a la contribución de la herencia al desarrollo de la EPOC5-15. Los factores genéticos pueden modular el impacto del humo del tabaco sobre el pulmón. De este modo, serán clave para determinar la velocidad de deterioro de la función pulmonar y condicionarán en parte qué pacientes desarrollarán la EPOC y su gravedad. Estas afirmaciones están basadas en estudios de casos y controles que han mostrado un aumento de la prevalencia de EPOC en los familiares de los casos en comparación con los familiares de los controles, lo cual no puede explicarse por otros factores de riesgo conocidos5-7. Hay también una mayor prevalencia de alteración de las pruebas funcionales respiratorias entre los hijos de pacientes con EPOC que entre sus cónyuges5,8-15. Estudios transversales han mostrado una disminución de la prevalencia de la enfermedad y menos similitud en la función pulmonar a medida que aumenta la distancia genética6,13,16. Aunque los resultados de varios de estos estudios muestran una agrupación de los casos de EPOC en familias, no hay un claro patrón mendeliano de herencia.
Entre los factores genéticos que estarían implicados en el desarrollo de EPOC, el ejemplo más claro lo constituye el déficit de la antiproteasa alfa-1-antitripsina (AAT), que comporta un riesgo muy aumentado de desarrollar enfisema a una edad temprana, sobre todo en fumadores17. En esta revisión tratamos de destacar los aspectos clínicos más relevantes de esta deficiencia y, sobre todo, la importancia de realizar un diagnóstico precoz, ya que es una enfermedad en la que pueden adoptarse medidas para frenar la evolución del enfisema, entre ellas, la administración de un tratamiento sustitutivo con AAT procedente de plasma de donantes en los individuos en que esté indicado.
Variantes de la alfa-1-antitripsina
El descubrimiento de Laurell y Erikson18 de que los pacientes con concentraciones plasmáticas bajas de globulinas alfa tenían un aumento de la prevalencia de enfisema fue la primera evidencia de un factor de riesgo genético para el desarrollo de la EPOC. La AAT es una potente antiproteasa y una de las pocas enzimas que pueden inhibir la elastasa del neutrófilo, una importante enzima en la patogenia y el desarrollo de la EPOC. La AAT se sintetiza fundamentalmente en el hígado, aunque también se produce en los macrófagos alveolares y en los monocitos de sangre periférica19. Es una proteína muy polimorfa, con más de 75 variantes identificadas por métodos de electroforesis y de isoelectroenfoque20-22. La mayoría de estas variantes carece de significado clínico, pero existen unas 30 que pueden tener repercusiones patológicas. Las variantes se clasifican de acuerdo con su velocidad de migración electroforética en un campo magnético: se denomina M (medium) a las de velocidad media, F (fast) a las de velocidad rápida y S (slow) a las de migración lenta. Cuando se fueron descubriendo otras variantes, se designaron con las letras iniciales del alfabeto a las anódicas, y con las finales a las catódicas.
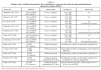
Es útil, desde un punto de vista clínico, clasificar las variantes de la AAT en 3 categorías (fig. 1):
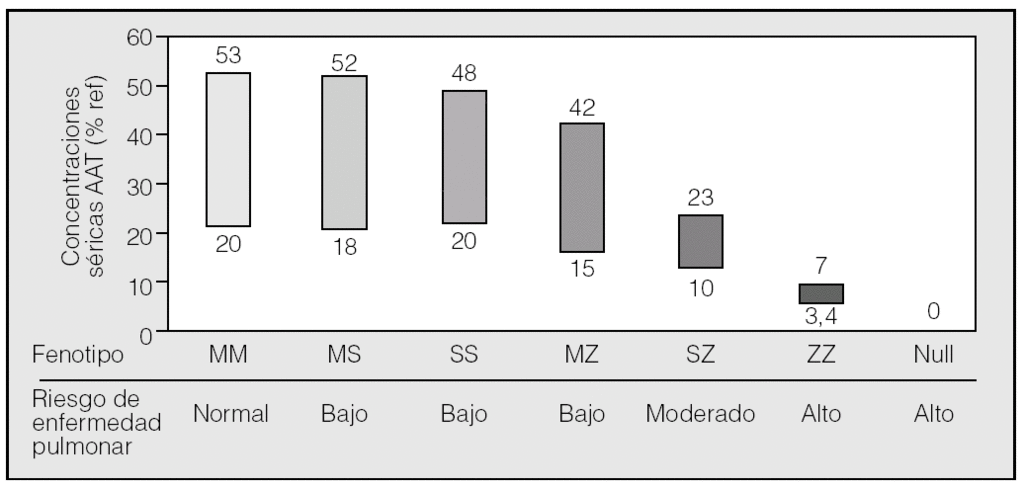
Fig. 1. Intervalos de concentraciones séricas de alfa-1-antitripsina (AAT) según fenotipo. En el eje de abscisas se representan los fenotipos PI más frecuentes y en el de ordenadas, las concentraciones plasmáticas que expresa cada fenotipo, en porcentaje del valor de referencia. El tamaño de las barras está en relación con la prevalencia de cada fenotipo; el valor de corte para protección es del 35%. Por debajo de este valor están todos los PIZZ y un tercio de los PISZ. Hay un gran solapamiento de valores de los fenotipos PIMS, PIMZ y PISS.
1. Normales, caracterizadas por concentraciones plasmáticas de AAT en un intervalo normal y que no se asocian con un aumento del riesgo para el desarrollo de enfermedad pulmonar o hepática. Esta categoría incluye fundamentalmente la variante genotípica M y sus subvariantes.
2. Deficitarias, que se caracterizan por presentar concentraciones plasmáticas bajas pero no indetectables de AAT. En este grupo se incluyen fundamentalmente las variantes S y Z. La variante deficitaria Z expresa aproximadamente un 10-20% de AAT y la variante S, más común en el área mediterránea, expresa aproximadamente el 50-60% de AAT.
3. Nulas, con concentraciones indetectables de AAT en plasma y asociadas con un aumento del riesgo para el desarrollo de enfisema.
La mayor parte de los casos de déficit de AAT están causados por el estado homocigoto para el alelo deficitario PIZ o por combinaciones de los 2 alelos deficitarios más frecuentes (PIS y PIZ).
La variante Z de la AAT (342 Glut-Lys) da lugar a una proteína con una función antiproteolítica deficiente, pero sobre todo lo que condiciona las concentraciones deficitarias es que es una proteína mal procesada en el retículo endoplásmico rugoso hepático, ya que polimeriza y se agrega en el interior de los hepatocitos, donde puede provocar enfermedad hepática23.
La variante S (264 Glu-Val) se encuentra en cerca del 25% de los individuos de poblaciones del sur de Europa24,25 y en estado homocigoto da lugar a concentraciones de AAT del 60% de los valores normales. Este estado homocigoto no se ha demostrado que se asocie con aumento del riesgo para el desarrollo de EPOC.
Los individuos con la rara combinación alélica SZ son menos frecuentes que los heterocigotos MS o MZ y tienen concentraciones de AAT de aproximadamente un 40% de los valores normales. El riesgo de desarrollo de enfermedad pulmonar en estos pacientes se ha investigado en diversos estudios con diferentes métodos. En estudios de casos y controles no se ha encontrado una mayor proporción de individuos con el fenotipo SZ entre los pacientes con EPOC frente a la encontrada en un grupo control26,27. En contraste, otros estudios de base poblacional han observado un aumento del riesgo del desarrollo de EPOC en estos individuos si son fumadores28-30, con tasas de obstrucción similares a las de los individuos homocigotos PIZ, mientras que este riesgo no se demostró en los no fumadores. Por último, un metaanálisis publicado recientemente31 y realizado sobre 6 estudios ha demostrado un aumento del riesgo para el desarrollo de EPOC en los individuos con la combinación SZ, con una odds ratio de 3,26 (intervalo de confianza del 95%, 1,24-8,57). Sin embargo, debido al limitado número de individuos SZ con información sobre la exposición tabáquica, los autores no han podido calcular la odds ratio por separado para los individuos fumadores y los no fumadores.
En conclusión, son varios los estudios que han destacado el tabaquismo como el mayor factor de riesgo para que los individuos con fenotipo SZ desarrollen enfisema pulmonar, mientras que el riesgo en no fumadores parece no ser diferente del de la población normal.
Además de las variantes deficitarias más frecuentes, mediante técnicas de secuenciación del ADN se han identificado otras variantes alélicas deficitarias. Se han descrito 8 variantes de la familia de alelos P, algunas de las cuales sólo pueden diferenciarse mediante la técnica de determinación del fenotipo por isoelectroenfoque y otras requieren además estudio del genotipo al presentar el mismo punto isoeléctrico32,33. De entre ellas, únicamente la PI Plovel y la PI Pduarte se han asociado con concentraciones bajas de AAT y enfisema pulmonar32-34. En un estudio realizado en España se detectó el alelo PI Plovel en miembros de 5 familias de diferentes áreas geográficas, lo que hace pensar que la prevalencia de este alelo puede ser mayor de lo esperado inicialmente. Probablemente la no detección previa de este alelo podría estar relacionada con una interpretación incorrecta de los patrones de isoelectroenfoque35.
La variante alélica PI Mpalermo se ha descrito en varias familias del área mediterránea35,36 y se caracteriza por tener una deleción de 3 pares de bases en las posiciones que codifican para Phe51 o Phe52, y por presentar una concentración de AAT similar a la del alelo PIZ.
Otra variante deficitaria poco frecuente resulta de la sustitución de una Pro en posición 369 por una Ser, y da lugar a la variante denominada Mvall d'hebron, que fue caracterizada por primera vez en nuestro país37.
Otra nueva variante identificada en España, denominada PI Ybarcelona, se caracteriza por sustituciones en las posiciones Asp256 en el exón III y Pro391 en el exón V38. Esta variante en estado homocigoto da lugar a un déficit grave de AAT, con concentraciones muy bajas de AAT y clínicamente indistinguible del déficit grave PIZZ, ya que cursa con enfisema pulmonar de aparición precoz. Los individuos con esta variante en estado heterocigoto presentan concentraciones intermedias de AAT y un riesgo de enfermedad pulmonar similar al de los individuos MZ39.
Variante deficitaria Z
Los individuos con la variante deficitaria Z en estado homocigoto tienen concentraciones de AAT circulante inferiores al 15% de lo normal y una tasa acelerada de deterioro de la función pulmonar, incluso en ausencia de tabaquismo40,41. Sin embargo, el enfisema pulmonar se desarrolla a una edad más temprana entre los individuos que además son fumadores42,43. Las bajas concentraciones plasmáticas y tisulares de AAT son insuficientes para proteger el tejido conectivo del pulmón de la acción de las proteasas del neutrófilo.
Aunque el fenotipo PIZZ es indudablemente un factor de riesgo genético para el desarrollo de EPOC, hay una variación considerable en la expresión clínica del déficit44,45. Esta variabilidad no es enteramente atribuible a la diferencia en la exposición al humo del tabaco, dado que la tasa de deterioro de la función pulmonar en individuos ZZ que no son fumadores es también muy variable41.
Es posible que otras mutaciones puedan actuar como modificadoras del curso clínico de los individuos homocigotos ZZ. Un estudio ha relacionado un polimorfismo (C774T) en el gen de la sintetasa del óxido nítrico endotelial con el riesgo para el desarrollo de EPOC en los individuos ZZ. El óxido nítrico puede desempeñar un papel en la patogenia de la EPOC porque regula el tono vascular y de la vía aérea en el pulmón y facilita la adhesión de los leucocitos al endotelio. El alelo 774T es significativamente más frecuente en los individuos ZZ cuyo volumen espiratorio forzado en el primer segundo (FEV1) es menor del 35% del predicho que en los sujetos ZZ cuyo FEV1 es mayor del 35%46.
Otro polimorfismo modificador de la expresión clínica del déficit de AAT es el de la glutatión S-transferasa P1, una subfamilia de la glutatión S-transferasa que se expresa ampliamente en todo tipo de células epiteliales, incluidas las pulmonares47,48, y que participa en la destoxificación de sustancias electrofílicas y productos del estrés oxidativo derivados del humo de tabaco49. El polimorfismo que da lugar a una sustitución de una isoleucina (Ile) por una valina (Val) en posición 105 puede alterar la actividad de dicha enzima, ya que esta posición está muy cercana al sitio de fijación de la enzima a los sustratos sobre los que actúa. Un estudio de casos y controles ha demostrado que la presencia en individuos fumadores con déficit de AAT de al menos un alelo Val en esta posición 105 confiere un aumento del riesgo para el deterioro de la función pulmonar. Sin embargo, los polimorfismos en este gen no parecen influir en la gravedad del deterioro de la función pulmonar en pacientes con EPOC y con concentraciones normales de AAT. Esto parece indicar que el papel protector de la enzima en su variente homocigota Ile105 es de menor magnitud que el debido a la AAT, por lo que su sustitución no tendría repercusión clínica en fumadores con concentraciones normales de AAT50.
Epidemiología del déficit de alfa-1-antitripsina
El alelo Z de la AAT predomina en poblaciones del norte de Europa y es más raro en poblaciones del sur de Europa, asiáticos y población de raza negra51. En España se ha observado una frecuencia génica para el alelo Z del 1,5% en la población general24,25, de modo que para una población aproximada de 40 millones de personas podrían esperarse 8.000 pacientes con la forma deficitaria grave homocigota PIZZ. En un estudio realizado en España se calculó el número de individuos de los 5 fenotipos deficientes más frecuentes (PIMS, PIMZ, PISS, PISZ y PIZZ), a partir de resultados de los principales estudios epidemiológicos publicados sobre la frecuencia del déficit de AAT. De acuerdo con estos cálculos, podría haber más de 9 millones de individuos portadores de estos alelos, de los cuales el 80% sería fenotipo PIMS y el resto sería PIMZ (13%), PISS (4,7%), PISZ (1,6%) y PIZZ (0,1%). De acuerdo con las estimaciones sobre penetrancia del fenotipo PIZZ, se calculó que podría haber 2.526 adultos con EPOC y 4.030 individuos (niños y adultos) con hepatopatías crónicas relacionadas con este fenotipo52.
La necesidad de disponer de unos criterios unificados en todo el país para instaurar el tratamiento sustitutivo y para su seguimiento impulsó la creación en 1993 del Registro Español de Pacientes con Déficit de AAT. Sus objetivos iniciales fueron: conocer las características y la frecuencia del déficit de AAT en España, establecer normativas sobre el tratamiento y el seguimiento de pacientes con el déficit, ofrecer información a los médicos que tratan a estos pacientes, incrementar el conocimiento y el interés por esta enfermedad e intentar disminuir el infradiagnóstico del déficit. Desde su origen, dicho registro es un grupo de trabajo del Área IRTS (Insuficiencia Respiratoria y Trastornos del Sueño) de la Sociedad Española de Neumología y Cirugía Torácica (SEPAR). La organización la componen 2 coordinadores, un comité asesor y 87 centros participantes distribuidos por toda España y Andorra53.
La prevalencia del déficit de AAT, calculada a partir de los datos de frecuencia génica en España, contrastan con la realidad del Registro Español de pacientes, que desde su fundación en el año 1993 incluye a unos 410 pacientes de 16 de las 17 comunidades autónomas53-55. A pesar de que la tasa de detección del déficit en España es similar, o incluso superior, a la de otros países de nuestro entorno56, todavía sigue siendo baja con respecto al número estimado de pacientes.
El Registro Español reúne información acerca de la evolución funcional de los pacientes registrados, la frecuencia del tratamiento sustitutivo y la posible aparición de efectos adversos con este tratamiento. El comité asesor se reúne periódicamente para evaluar y analizar la evolución de la base de datos del registro y actualizar las normativas referentes al tratamiento y al seguimiento. Del mismo modo, este comité organiza una reunión anual abierta en el marco del congreso nacional de la SEPAR. En 1999 el Registro Español de Pacientes con Déficit de AAT se unió al Registro Internacional de esta enfermedad (AIR), que incluye a pacientes de 20 países de 4 continentes57, y desde el año 2002 el Registro Español dispone de un nuevo sistema on-line de recogida de datos dentro de la página web oficial de la SEPAR, de forma que médicos de toda España pueden introducir los datos de los pacientes a través de Internet (www.separ.es/air)
Riesgo de enfisema en portadores heterocigotos del déficit de alfa-1-antitripsina (MZ, MS)
La pregunta de si el déficit intermedio de AAT es un factor de riesgo para el desarrollo de la EPOC ha generado una gran controversia. Este déficit está causado sobre todo por los fenotipos MS y MZ, presentes en las poblaciones caucásicas en aproximadamente un 10 y un 3%, respectivamente. En España, según los resultados del cálculo de individuos con los 5 fenotipos deficientes más frecuentes (PIMS, PIMZ, PISS, PISZ y PIZZ), podría haber más de 9 millones de individuos portadores de estos fenotipos52.
Se han realizado estudios de casos y controles donde se han comparado las prevalencias de los fenotipos que dan lugar a concentraciones intermedias en pacientes con EPOC frente a las encontradas en individuos sin obstrucción al flujo aéreo. Los resultados de muchos de estos estudios han demostrado un aumento de la prevalencia de heterocigotos MZ entre los pacientes con EPOC frente al grupo control, con una odds ratio para la forma MZ que oscila entre 1,5 y 558.
Hay también estudios sobre el riesgo asociado al fenotipo MZ realizados sobre individuos de la población general. En ellos se determina su función pulmonar y se compara con la función pulmonar de los MM. Estos estudios serían menos susceptibles de error, dado que todos los individuos son reclutados del mismo modo y proceden de la misma población. Los resultados de la mayoría de estos estudios no han mostrado diferencias significativas en los síntomas respiratorios o en la función pulmonar de los MZ frente a los MM. Sin embargo, muchos de ellos están basados en muestras pequeñas y no todos los sujetos tienen la suficiente exposición tabáquica59-61. Otros estudios realizados sobre población general han demostrado diferencias entre individuos fumadores MZ e individuos MM en la función pulmonar o en la pérdida de retroceso elástico del parénquima pulmonar62.
En un estudio reciente realizado sobre población general, en el que se determinaron las concentraciones y el fenotipo de AAT y la tasa anual del deterioro del FEV1, con un seguimiento medio de 21 años, se ha demostrado un deterioro del FEV1 es un 19% mayor entre los individuos MZ que entre los MM (25 frente a 21 ml; p = 0,048). Además, la tasa de individuos con obstrucción al flujo aéreo fue del 19% en el grupo con el fenotipo MZ, frente el 15% encontrado en el grupo con el fenotipo MM (p = 0, 023)63.
En conclusión, los resultados de los estudios de casos y controles y algunos estudios realizados sobre la población general han indicado que el genotipo MZ es un factor de riesgo para el desarrollo de EPOC, aunque el aumento del riesgo es probablemente bastante modesto y sólo en fumadores. Los resultados más relevantes sobre el riesgo de enfermedad pulmonar asociada a heterocigotos se exponen en la tabla I26,59-69.
En cuanto a los fenotipos PIMS y PISS, los datos disponibles sobre riesgo de desarrollar enfermedades asociado a estos fenotipos son conflictivos y poco concluyentes, por lo que la mayoría de los autores considera que estos fenotipos no suponen un riesgo incrementado de desarrollar enfermedad pulmonar70.
Se han realizado varios estudios con el fin de investigar la asociación del déficit intermedio de AAT con otras enfermedades pulmonares y fundamentalmente el asma bronquial. Varios de ellos han demostrado cómo la presencia del alelo Z en estado heterocigoto (PIMZ) da lugar a una mayor gravedad del asma en niños y adolescentes71,72. Un estudio realizado en población adulta analizó la distribución de los fenotipos deficitarios de AAT en pacientes asmáticos y no encontró diferencias en esta distribución frente a la encontrada en la población general. Además, los fenotipos deficitarios en estado heterocigoto no implicaron una mayor gravedad o una expresión clínica diferente del asma en adultos73.
Detección de casos de déficit de alfa-1-antitripsina, ¿por qué, cuándo, cómo?
Los estudios poblacionales realizados en España indican que el déficit de AAT es una enfermedad infradiagnosticada y con frecuentes retrasos en el diagnóstico54,55,74.
Entre todas las causas de infradiagnóstico destaca el que se atribuya el enfisema únicamente al tabaco, sin investigar la posibilidad del déficit de AAT. Hoy día todavía se considera en algunas normativas que sólo se debe sospechar el déficit en pacientes con enfisema grave, jóvenes y/o no fumadores75; sin embargo, estos individuos son sólo una pequeña parte de todos los casos y muchos pacientes no encajan en estos parámetros de gravedad45. En un estudio realizado en España, el retraso en el diagnóstico, medido como el tiempo entre el diagnóstico de la EPOC y el del déficit de AAT, fue de un promedio de 10 años76.
La importancia del diagnóstico precoz se basa en la posibilidad de realizar un programa enérgico de abandono del hábito tabáquico y de tratamiento de la enfermedad pulmonar, un estudio familiar que puede identificar a individuos de riesgo elevado en fases más tempranas y la posibilidad de iniciar un tratamiento sustitutivo con AAT en los individuos que cumplen los criterios establecidos.
Las técnicas más frecuentemente usadas hasta hoy para el diagnóstico de laboratorio del déficit de AAT incluyen la determinación de las concentraciones séricas de AAT y la identificación del fenotipo de la AAT por enfoque isoeléctrico a un pH de 4,2-4,977,78. En estudios previos se ha evaluado la eficacia de métodos para el cribado del déficit de AAT como la cuantificación de la banda α1 del proteinograma. Los pacientes con un déficit grave de AAT (fenotipo PIZZ) presentaban de forma constante unas concentraciones de globulinas α1 por debajo de los límites de la normalidad establecidos78. Sin embargo, debido a la escasa prevalencia del déficit de AAT y a las diversas causas que pueden provocar un descenso en las proteínas séricas, unas concentraciones bajas de la banda α1 no siempre son indicativas de un déficit de AAT. En estas circunstancias es obligado confirmarlo mediante la cuantificación de AAT sérica por nefelometría y, en caso de obtenerse valores disminuidos, determinar el fenotipo PI.
La Organización Mundial de la Salud recomendó en 1997 la determinación de las concentraciones séricas de AAT en todos los pacientes con EPOC. A los pacientes con resultados anómalos en este cribado se les debería realizar la determinación del fenotipo79. La American Thoracic Society y la European Respiratory Society han elaborado un documento conjunto de consenso para el diagnóstico y tratamiento del déficit de AAT y recomiendan realizar pruebas de diagnóstico de este déficit a todos los adultos con enfisema pulmonar o EPOC (recomendación tipo A)80.
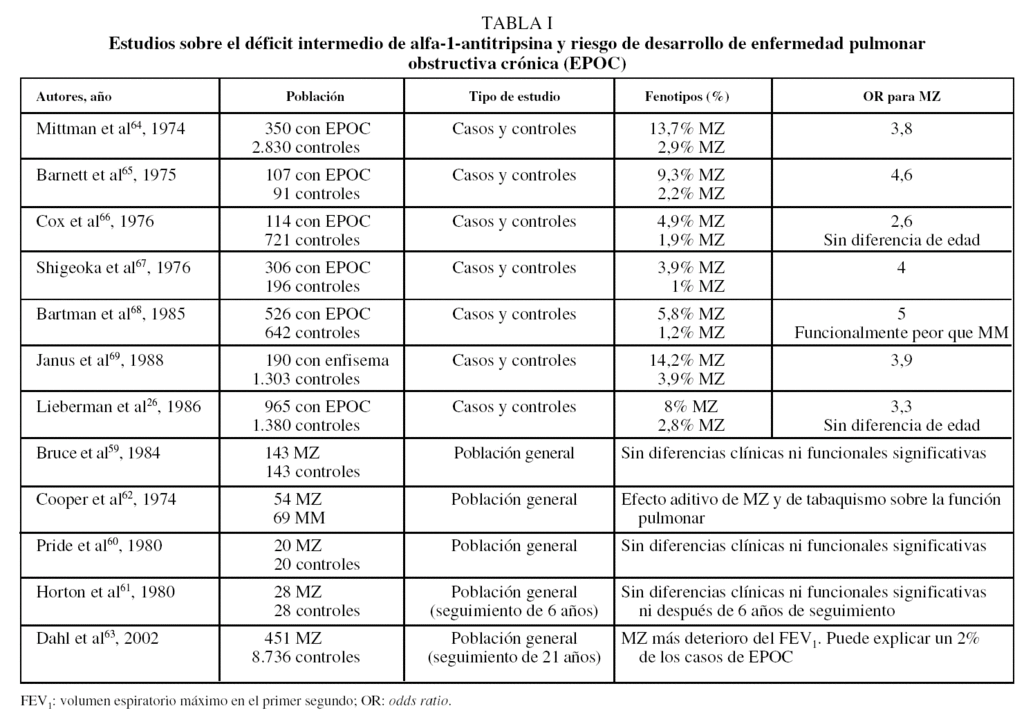
Siguiendo estas recomendaciones, diversos países han iniciado programas de detección de casos (tabla II)81-84. En Italia se ha aplicado el cribado mediante fenotipificación con técnicas de isoelectroenfoque a individuos con elevada sospecha clínica o en estudios familiares de déficit de AAT, con lo que el rendimiento diagnóstico ha sido elevado: 151 casos detectados entre 1.841 muestras analizadas (8,2%), de los cuales 118 fueron ZZ (78%)81.
Por el contrario, en otro programa realizado en Alemania82 se han observado tasas de detección muy inferiores, de modo que entre los 1.060 individuos estudiados no se ha detectado ninguno con el genotipo deficitario Z en estado homocigoto. Esto muy posiblemente se deba a que la población estudiada estaba constituida por pacientes con todo tipo de enfermedades respiratorias crónicas (asma, bronquiectasias y EPOC), independientemente del grado de sospecha del déficit de AAT.
Otro estudio realizado en EE.UU. ha aplicado el programa detección en 969 pacientes con diagnósticos de enfisema, asma o bronquitis crónica y las tasas de detección fueron de un caso ZZ por cada 31 muestras, y una de cada 9 muestras era heterocigoto MZ83.
Estos programas son posibles gracias al desarrollo de técnicas precisas para cuantificar las concentraciones de AAT y determinar el genotipo a partir de muestras de sangre capilar en papel secante (fig. 2). Estudios previos en España han validado un método específico de inmunonefelometría para la cuantificación de AAT en muestras de gota de sangre en papel secante85. También se ha utilizado en la detección de los genotipos deficitarios más frecuentes del déficit de AAT, donde ha demostrado ser coste-efectivo y aplicable en programas de detección de casos de este déficit a gran escala86. Estas muestras requieren una pequeña cantidad de sangre, que se obtiene con una extracción de sangre capilar del pulpejo del dedo y que posteriormente se coloca sobre el papel secante, para proceder posteriormente al envío de la muestra por correo convencional.
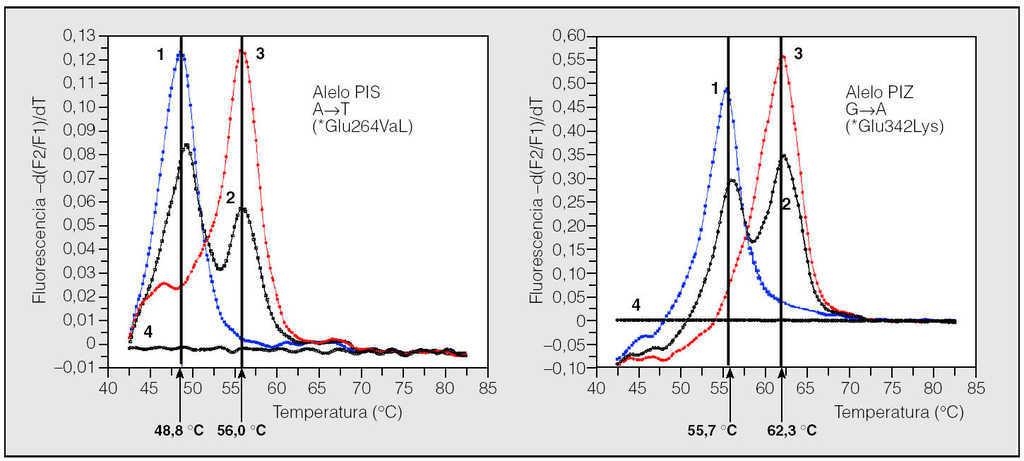
Fig. 2. Resultados de la genotipificación del déficit de alfa-1-antitripsina (AAT) en gotas de sangre seca mediante el analizador LightCycler. Curvas del genotipo PIS en el LightCycler: caso 1, homocigoto PISS; caso 2, heterocigoto PIMS; caso 3, homocigoto PIMM, y caso 4, negativo. Curvas del genotipo PIZ en el LightCycler: caso 1, homocigoto PIZZ; caso 2, heterocigoto PIMZ; caso 3, homocigoto PIMM, y caso 4, negativo. A: adenina; T: timina; Glu: ácido glutámico; Val: valina; G: guanina; Lys: lisina. Adaptada de Rodríguez82.
En España este procedimiento se está realizando como una iniciativa del Registro Español de Pacientes con Déficit de AAT (fig. 3). En una primera fase se ha comprobado el correcto funcionamiento del circuito utilizado para la obtención y envío de las muestras al laboratorio central. Además, se ha demostrado que es un método aplicable, cómodo y bien aceptado por los médicos participantes y permite cuantificar la concentración sérica de AAT y la detección del alelo Z87.
Fig. 3. Cuestionario de recogida de datos de cada paciente junto a los discos de papel secante empleados en el programa de cribado del déficit de alfa-1-antitripsina.
Hasta el momento actual se han analizado muestras de 2.138 pacientes con EPOC y se han detectado 8 casos de individuos homocigotos PIZZ84, lo que ofrece una tasa de diagnósticos intermedia a las de las experiencias en Italia y Alemania. A la hora de diseñar un programa de detección de casos de déficit de AAT, se deben tener en cuenta tanto el protocolo de procesamiento de las muestras como los criterios de inclusión de los candidatos, ya que ambos factores van a influir decisivamente en el rendimiento del programa y en su coste.
A principios de 2005 el Registro Español inició el proyecto IDDEA (Información y Detección del Déficit de Alfa-1-Antitripsina) con el fin de poner el diagnóstico del déficit al alcance del mayor número de médicos que tratan pacientes con EPOC. Este programa informa sobre la importancia del diagnóstico temprano según las normativas de la Organización Mundial de la Salud y la American Thoracic Society/European Respiratory Society, además de proporcionar los medios para la detección del déficit de AAT en sus pacientes. Tras el diagnóstico y la inclusión de los pacientes en el Registro Español, se puede comprobar si cumplen los criterios para iniciar un tratamiento sustitutivo y decidir su indicación88.
La detección del déficit de AAT es crucial para detectar nuevos casos en un país como España, donde la enfermedad tiene una baja prevalencia y, como consecuencia, hay un bajo índice de sospecha de esta entidad en pacientes con EPOC.
Trabajo financiado por la Red Respira RTIC 03/11 ISCII.
Correspondencia: Dr. C. de la Roza.
Servicio de Neumología. Hospital Clínic.
Villarroel, 170. 08036 Barcelona. España.
Correo electrónico: croza@clinic.ub.es
Recibido: 27-7-2005; aceptado para su publicación el 6-9-2005.







