
Alpha-1 antitrypsin (AAT) is the most abundant protease inhibitor of human serum, with average concentrations in healthy individuals ranging between 120 and 220mg/dl determined by nephelometry. The synthesis of AAT depends on the SERPINA1 gene.1,2
The most common deficiency alleles found in Spain are the PI*S and the PI*Z, and approximately 1/5 and 1/61 individuals in Spain, respectively, are carriers of this mutation.3–5 These alleles have been associated with the development of pulmonary emphysema in early adulthood, liver diseases in children and adults, and other entities such as systemic vasculitis (especially Wegener's granulomatosis positive for c-ANCA) or necrotizing panniculitis.2,6
Few population genetic studies of AAT deficiency alleles have been conducted in the Canary Islands,7 and none have identified rare deficiency alleles. For this reason, the aim of this study was to describe the spectrum of mutations found in the population of Tenerife seen at the Hospital Universitario Nuestra Señora de Candelaria (HUNSC).
This was a cross-sectional, descriptive study of a series that includes index cases with AAT plasma levels below 100mg/dl who were referred to the Clinical Laboratory department of the HUNSC on clinical suspicion of alpha-1 antitrypsin deficiency (AATD) between January 1, 2009 and August 31, 2016.
AAT serum values were determined by nephelometry (BN ProSpec System, Siemens). Genomic DNA was purified from whole blood with EDTA (QIAamp DNA blood mini kit, Qiagen). The two most common deficiency alleles PI*S and Pi*Z were genotyped by real-time PCR using FRET probes (LightCycler 2.0). The genotypes obtained were correlated with previously determined plasma AAT values, following the routine protocol in our hospital.8 Patients with AAT plasma levels out of range of those expected for the genotypes S/Z obtained underwent complete sequencing of the coding exons and intronic sequences flanking the SERPINA1 gene (BigDye v3.1, Thermo Fisher), replacing conventional isoelectric focusing phenotyping. SERPINA1 sequencing was performed in a AB3500 capillary sequencer (Applied Biosystems), and results were compared with the NM_001127701.1 reference sequence (SeqScape 3.0).
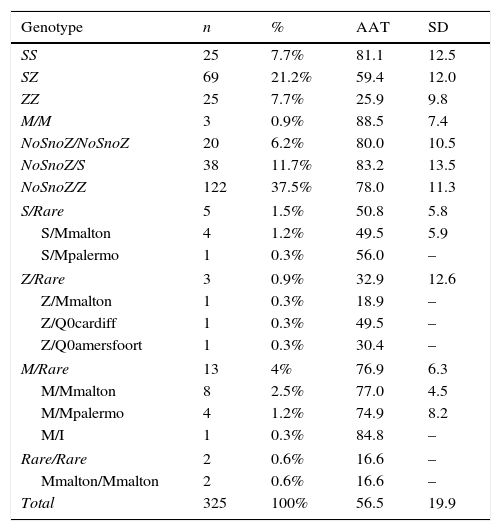
In total, 325 AATD patients with AAT plasma levels <100mg/dl were included in the study. Studies directed at the PI*S and Pi*Z deficiency alleles were conducted in all patients. Genotypes are shown in Table 1.
Genotypes and AAT Levels Obtained in the Study.
| Genotype | n | % | AAT | SD |
|---|---|---|---|---|
| SS | 25 | 7.7% | 81.1 | 12.5 |
| SZ | 69 | 21.2% | 59.4 | 12.0 |
| ZZ | 25 | 7.7% | 25.9 | 9.8 |
| M/M | 3 | 0.9% | 88.5 | 7.4 |
| NoSnoZ/NoSnoZ | 20 | 6.2% | 80.0 | 10.5 |
| NoSnoZ/S | 38 | 11.7% | 83.2 | 13.5 |
| NoSnoZ/Z | 122 | 37.5% | 78.0 | 11.3 |
| S/Rare | 5 | 1.5% | 50.8 | 5.8 |
| S/Mmalton | 4 | 1.2% | 49.5 | 5.9 |
| S/Mpalermo | 1 | 0.3% | 56.0 | – |
| Z/Rare | 3 | 0.9% | 32.9 | 12.6 |
| Z/Mmalton | 1 | 0.3% | 18.9 | – |
| Z/Q0cardiff | 1 | 0.3% | 49.5 | – |
| Z/Q0amersfoort | 1 | 0.3% | 30.4 | – |
| M/Rare | 13 | 4% | 76.9 | 6.3 |
| M/Mmalton | 8 | 2.5% | 77.0 | 4.5 |
| M/Mpalermo | 4 | 1.2% | 74.9 | 8.2 |
| M/I | 1 | 0.3% | 84.8 | – |
| Rare/Rare | 2 | 0.6% | 16.6 | – |
| Mmalton/Mmalton | 2 | 0.6% | 16.6 | – |
| Total | 325 | 100% | 56.5 | 19.9 |
AAT: alpha-1 antitrypsin deficiency; SD: standard deviation; rare: rare deficiency variants.
Forty-six patients (14.2%) had a genotype associated with severe deficiency (AAT<50mg/dl), of which 24 were PI*ZZ, 10 PI*SZ, 1 PI*SS, 2 PI*S/Mmalton, 1 PI*Z/Q0amersfoort, 1 PI*Z/Q0cardiff, 1 PI*Z/Mmalton, 2 PI*Mmalton/Mmalton, 1 PI*MZ, 2 PI*S/NoSnoZ and 1 PI*Z/NoSnoZ. In total, 279 patients (85.8%) were recorded as having mild-to-moderate deficiency (AAT between 50 and 100mg/dl), distributed as follows: 119 PI*Z/NoSnoZ, 59 PI*SZ, 36 PI*S/NoSnoZ, 24 PI*SS, 8 PI*M/Mmalton, 4 PI*M/Mpalermo, 2 PI*S/Mmalton, 1 PI*S/Mpalermo, 1 PI*ZZ, 1 PI*MZ, 1 PI*MI, 3 PI*MM and 20 NoSnoZ/NoSnoZ.
The percentage of carriers of rare deficiency alleles was 7.7%, 80% of whom had the mutation F76del (PI*Mmalton and PI*Mpalermo).
In this study, we performed an AATD diagnostic protocol based on the quantification of AAT plasma levels, followed by genotyping for deficiency alleles PI*S and PI*Z, as these are the most common in our population. In recent years, correlation of both results has been shown to be the most efficient technique, achieving an unequivocal diagnosis in approximately 96% of cases.8 In the remaining patients, SERPINA1 full-gene sequencing was performed, a technique that has been shown to improve the accuracy of AATD diagnosis.9 Most AATD study protocols use isoelectric focusing phenotyping as the gold standard. However, in our case, we decided to use SERPINA1 gene sequencing, due to the limitations of phenotyping in the identification of rare deficiency variants.10
An interesting finding in our study was the yield obtained from the combination of protein levels and PI*S and Pi*Z allele genotyping (84.9%), which was lower than reported in the literature.8 This low yield from the diagnostic protocol is due to the high prevalence of rare deficiency variants, particularly the mutation F76del (PI*Mmalton and PI*Mpalermo) that accounts for 81.5% of the rare variants encountered; this percentage is higher than described in similar studies conducted in Spain.11 Moreover, in the 25 patients who underwent SERPINA1 gene sequencing, we found 20 with NoSnoZ/NoSnoZ, 4 with PI*S/NoSnoZ, and 1 with genotype PI*Z/NoSnoZ with AAT plasma levels discordant with genotyping for the S and Z alleles. Although they were candidates for SERPINA1 full-gene sequencing, this was not performed as the technique was not available in our hospital at the time of the study. We should add that in 3 patients with mild-to-moderate AAT deficiency, no mutations were detected, despite SERPINA1 full-gene sequencing. This may be due to mutations in gene regulatory regions that were not studied,12 or mutations that are undetectable by sequencing that require alternative techniques for diagnosis.13
AATD is one of the most prevalent hereditary diseases in our population, and the mutation F76del, like the PI*Z allele, has been associated with the development of pulmonary emphysema and liver disease,14,15 so we believe it is appropriate to detect these mutations in all patients in whom it is impossible to obtain an unequivocal diagnosis with the standard genetic protocols combining serum AAT levels, genotyping for deficiency alleles PI*S and Pi*Z, and phenotyping.
Please cite this article as: Martínez Bugallo F, Figueira Gonçalves JM, Martín Martínez MD, Díaz Pérez D. Espectro de mutaciones deficitarias de alfa-1 antitripsina detectadas en Tenerife. Arch Bronconeumol. 2017;53:595–596.







