


Lymphomatoid granulomatosis (LG) is an uncommon entity, classified by the WHO among the group of B-cell lymphoproliferative syndromes associated with Epstein–Barr virus (EBV) infection.1 As lung is affected in more than 90% of cases, clinicians must establish a differential diagnosis against other diseases such as Wegener's granulomatosis, lymphoma or pulmonary metastases.
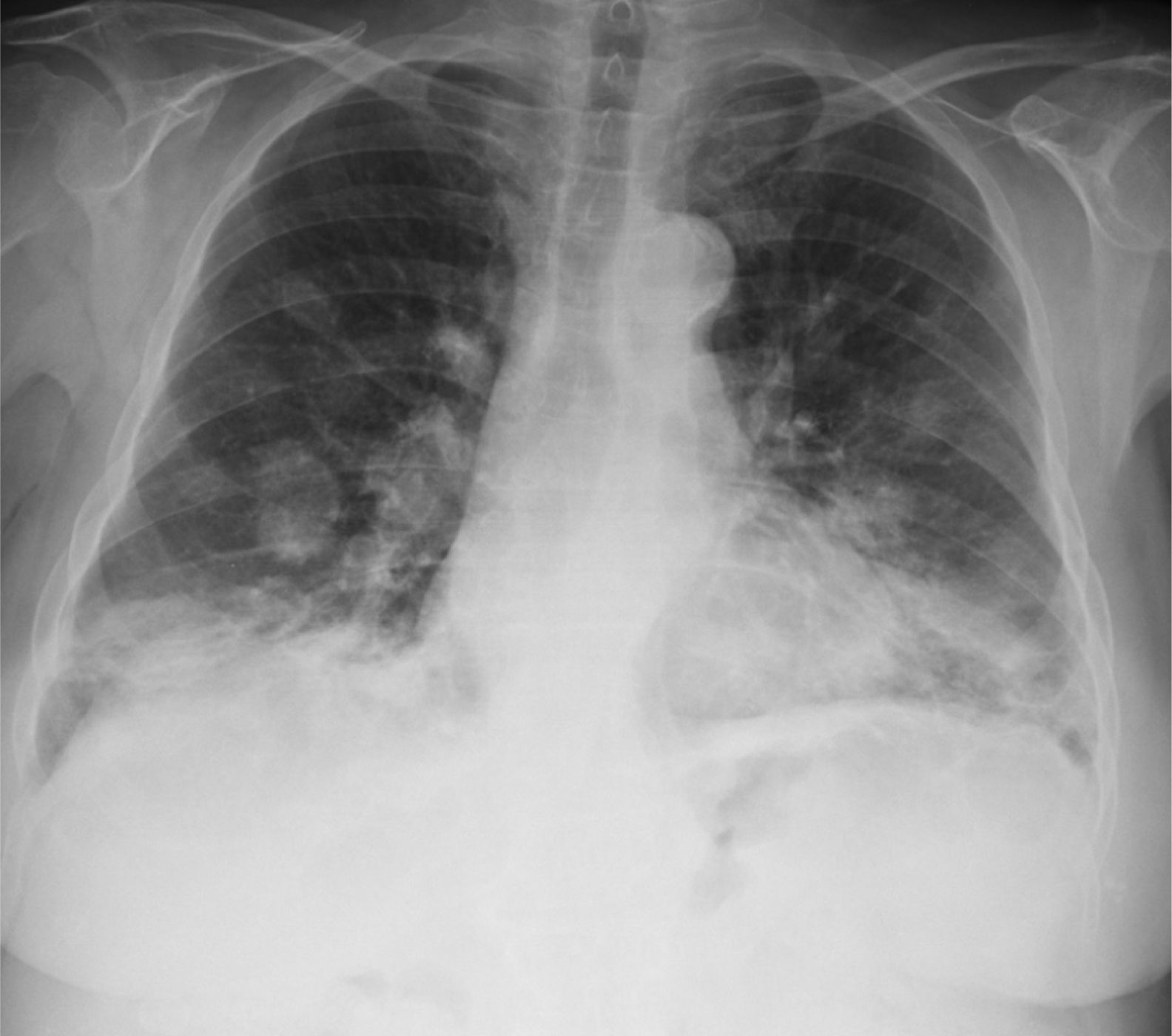
We report the case of a 76-year-old patient, with no toxic habits, hypertensive, with the chance finding on a chest X-ray of a “balloon release” image, finally diagnosed as LG. The patient was asymptomatic. No significant findings were observed on physical examination and lung auscultation was normal. Minimal leukocytosis was seen on clinical laboratory testing, which was normal for tumor markers. Imaging tests showed the presence of bilateral pulmonary nodules, predominantly in the lower lobes (Fig. 1), some with air bronchogram sign and occasional central cavities. Transbronchial and transthoracic biopsies were performed, but did not yield a diagnosis. An atypical surgical resection of a pulmonary node was performed, showing polymorphous lymphocytic infiltrate with an angiocentric and angiodestructive pattern, consisting of aberrant CD20 positive lymphoid cells on a background of small sized lymphocytes. An EBER probe was used to demonstrate the presence of EBV-infected cells, establishing a definitive diagnosis of grade 2 LG.2
In view of the radiological progression over the previous months, treatment was started with prednisone and IFN α-2b. Initial response was good, but after 18 months of treatment, the patient died from the disease.
LG generally occurs between the ages of 50 and 70 years, mainly in men.
It is one of the B-cell lymphoproliferative syndromes associated with EBV infection, and appears more frequently in immunosuppressed patients, and in association with the administration of azathioprine and methotrexate.2,3
The most frequently affected organ is the lung, but it can also affect the skin, the kidneys and the nervous system.
Patients may be asymptomatic at the time of diagnosis, but up to 60% can present non-specific symptoms such as cough, fever, rash, subcutaneous nodules, asthenia and anorexia, dyspnea, chest pain, ataxia or peripheral neuropathy.
Imaging tests typically show bilateral nodules and masses with peribronchovascular distribution, mainly in the lower lobes, which may converge or form central cavities. FDG uptake on PET is variable.4
Histopathological diagnosis is based on the triad of polymorphic lymphoid infiltrates, vasculitis, and focal necrosis.
The WHO proposes a histological classification according to the prominence of B-cells, from grade 1 to 3. The characteristics of these cells are similar to those of diffuse B-cell lymphoma.1
Treatment requires discontinuation of potentially causative medications, plus chemotherapy with IFN α-2b5 or rituximab.
The decision to treat must be based on the presence of symptoms, extensive involvement, or high-grade histopathological lesions.
Although rates of spontaneous remission of up to 20% have been reported, most cases progress, and mean survival is between 1 and 6 years.
Please cite this article as: Santalla-Martínez M, García-Quiroga H, Navarro-Menéndez I. Granulomatosis linfomatoide. Una entidad infrecuente a tener en cuenta en el diagnóstico diferencial de la imagen en suelta de globos. Arch Bronconeumol. 2015;51:606–607.







