

Within the spectrum of fibrosing interstitial lung diseases (f-ILDs) there is a subset of patients who exhibit a similar clinical course and prognosis to those with idiopathic pulmonary fibrosis (IPF), named progressive pulmonary fibrosis (PPF).1,2 The mechanisms underlying PPF are unclear. We hypothesize that the peripheral immune cell profile in PPF may differ from non-progressive f-ILDs (non-PPF). To test this hypothesis, we determined the blood immune cell profile in patients with f-ILDs at diagnosis and we explored their association with disease severity, as determined by lung function (forced vital capacity [FVC] and diffusing capacity for carbon monoxide [DLCO]). Patients were then followed-up for 12 months and classified post hoc as PPF or non-PPF. This allowed us to compare the relationship of the immune cell profile determined at baseline and PPF.
This observational study included 33 patients with f-ILDs, other than IPF. Diagnosis of ILDs was established through a multidisciplinary discussion based on clinical characteristics along with HRCT scan, bronchoalveolar lavage, and lung biopsy patterns if appropriate. Disease progression was defined as per international guidelines (ATS/ERS/JRS/ALAT), by ≥2 of the following criteria: worsening respiratory symptoms, lung function progression (absolute change in FVC ≥5% and/or DLCO ≥10%) and/or radiological progression.1 Lung function was measured according to international standards. Reference values were those of Roca et al.3 Peripheral blood was collected at diagnosis in EDTA tubes. Fluorescence activated cell staining and analysis (FACS) was used to profile B cells, T cells (and subpopulations), NK cells, NKT-like cells, monocytes, neutrophils, and eosinophils. Briefly, 120μl of blood was incubated with 30μl of the antibody mix during 30min at 4°C. Then, erythrocytes were lysed (BD FACS Lysing Solution, USA), and cells were incubated with the fixable Viability Stain (440UV, BD 566332) 15min at room temperature in the dark, washed and fixed using PFA 4%. Fixed samples data were acquired with a BD LSRFortessa 5 laser flow cytometer. FlowJo version 10 software (FlowJo LL, USA) was used for the analysis. The Ethics Committee of our institution approved the study (HCB/2017/0901), and all participants signed their informed consent. All statistics were computed with R version 3.6.2, using custom scripts.
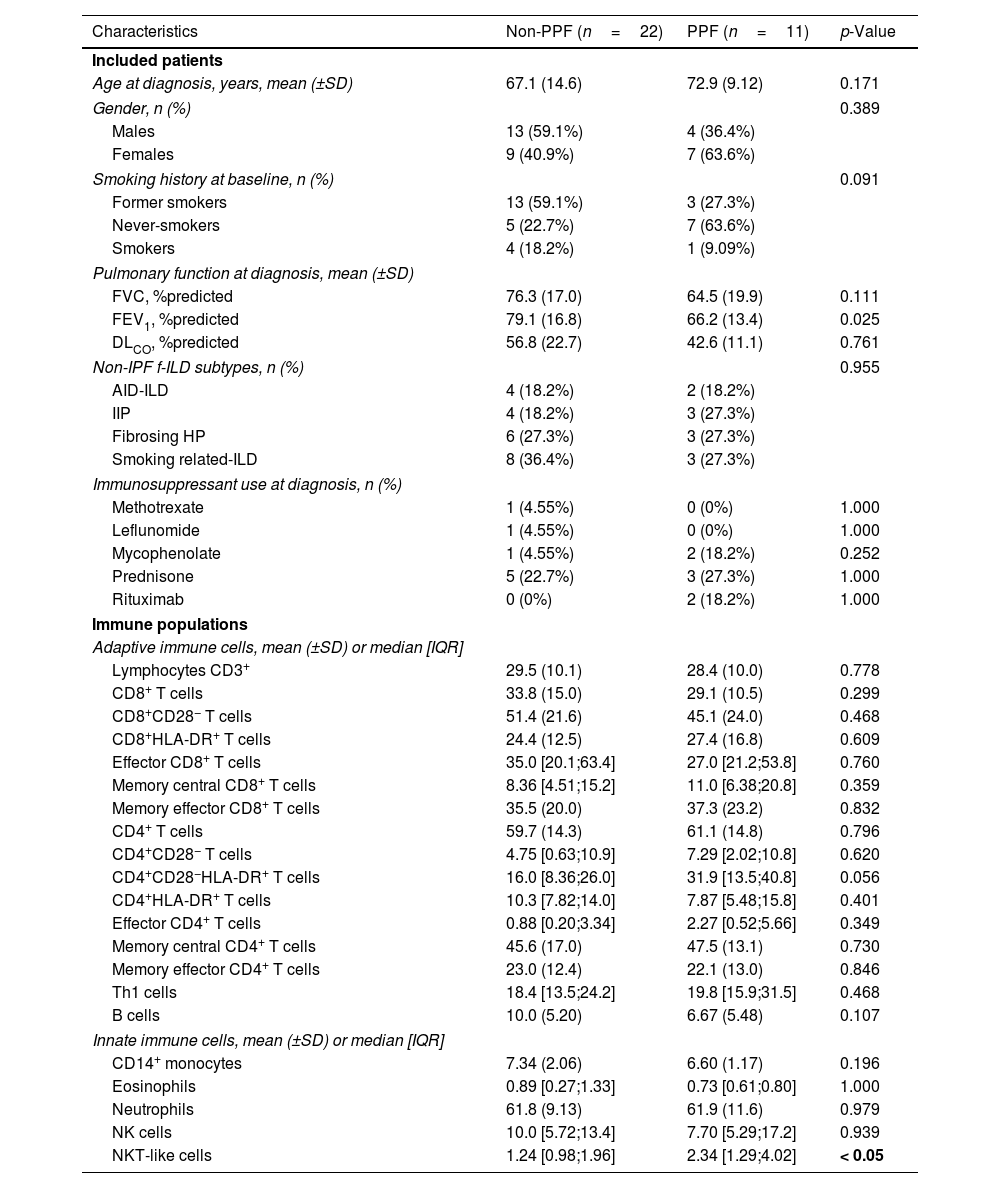
Eleven patients (33.3%) exhibited PPF during follow-up, whereas 22 (66.7%) remained stable. Table 1 presents the main demographic and clinical characteristics of these two groups at diagnosis, with further stratification by non-idiopathic pulmonary fibrosis subtypes available in Table S1. Age, gender, smoking status and proportion of f-ILD subtypes were similar across main groups. Most patients were naïve to immunosuppressive and antifibrotic treatment. In the entire population studied (n=33), we found significant correlations between several blood immune markers and lung function (Fig. 1A): FVC was negatively correlated with CD8+HLA-DR+ T cells and central memory (CM) CD4 T cells, whereas both FVC and DLCO were negatively related with the percentage of T helper type 1 (Th1) cells and CD4+CD28−HLA-DR+. We then explored which markers at diagnosis were associated with PPF. We found that PPF patients showed a significantly higher percentage of NKT-like cells (Fig. 1B and C), and a significant negative correlation between FVC and the percentage of CD8 T cells, which was not observed in non-PPF patients (Fig. 1D).
Characteristics of Participants and Immune Populations at Diagnosis in Fibrosing ILD Patients With and Without PPF During Follow-up.
| Characteristics | Non-PPF (n=22) | PPF (n=11) | p-Value |
|---|---|---|---|
| Included patients | |||
| Age at diagnosis, years, mean (±SD) | 67.1 (14.6) | 72.9 (9.12) | 0.171 |
| Gender, n (%) | 0.389 | ||
| Males | 13 (59.1%) | 4 (36.4%) | |
| Females | 9 (40.9%) | 7 (63.6%) | |
| Smoking history at baseline, n (%) | 0.091 | ||
| Former smokers | 13 (59.1%) | 3 (27.3%) | |
| Never-smokers | 5 (22.7%) | 7 (63.6%) | |
| Smokers | 4 (18.2%) | 1 (9.09%) | |
| Pulmonary function at diagnosis, mean (±SD) | |||
| FVC, %predicted | 76.3 (17.0) | 64.5 (19.9) | 0.111 |
| FEV1, %predicted | 79.1 (16.8) | 66.2 (13.4) | 0.025 |
| DLCO, %predicted | 56.8 (22.7) | 42.6 (11.1) | 0.761 |
| Non-IPF f-ILD subtypes, n (%) | 0.955 | ||
| AID-ILD | 4 (18.2%) | 2 (18.2%) | |
| IIP | 4 (18.2%) | 3 (27.3%) | |
| Fibrosing HP | 6 (27.3%) | 3 (27.3%) | |
| Smoking related-ILD | 8 (36.4%) | 3 (27.3%) | |
| Immunosuppressant use at diagnosis, n (%) | |||
| Methotrexate | 1 (4.55%) | 0 (0%) | 1.000 |
| Leflunomide | 1 (4.55%) | 0 (0%) | 1.000 |
| Mycophenolate | 1 (4.55%) | 2 (18.2%) | 0.252 |
| Prednisone | 5 (22.7%) | 3 (27.3%) | 1.000 |
| Rituximab | 0 (0%) | 2 (18.2%) | 1.000 |
| Immune populations | |||
| Adaptive immune cells, mean (±SD) or median [IQR] | |||
| Lymphocytes CD3+ | 29.5 (10.1) | 28.4 (10.0) | 0.778 |
| CD8+ T cells | 33.8 (15.0) | 29.1 (10.5) | 0.299 |
| CD8+CD28− T cells | 51.4 (21.6) | 45.1 (24.0) | 0.468 |
| CD8+HLA-DR+ T cells | 24.4 (12.5) | 27.4 (16.8) | 0.609 |
| Effector CD8+ T cells | 35.0 [20.1;63.4] | 27.0 [21.2;53.8] | 0.760 |
| Memory central CD8+ T cells | 8.36 [4.51;15.2] | 11.0 [6.38;20.8] | 0.359 |
| Memory effector CD8+ T cells | 35.5 (20.0) | 37.3 (23.2) | 0.832 |
| CD4+ T cells | 59.7 (14.3) | 61.1 (14.8) | 0.796 |
| CD4+CD28− T cells | 4.75 [0.63;10.9] | 7.29 [2.02;10.8] | 0.620 |
| CD4+CD28−HLA-DR+ T cells | 16.0 [8.36;26.0] | 31.9 [13.5;40.8] | 0.056 |
| CD4+HLA-DR+ T cells | 10.3 [7.82;14.0] | 7.87 [5.48;15.8] | 0.401 |
| Effector CD4+ T cells | 0.88 [0.20;3.34] | 2.27 [0.52;5.66] | 0.349 |
| Memory central CD4+ T cells | 45.6 (17.0) | 47.5 (13.1) | 0.730 |
| Memory effector CD4+ T cells | 23.0 (12.4) | 22.1 (13.0) | 0.846 |
| Th1 cells | 18.4 [13.5;24.2] | 19.8 [15.9;31.5] | 0.468 |
| B cells | 10.0 (5.20) | 6.67 (5.48) | 0.107 |
| Innate immune cells, mean (±SD) or median [IQR] | |||
| CD14+ monocytes | 7.34 (2.06) | 6.60 (1.17) | 0.196 |
| Eosinophils | 0.89 [0.27;1.33] | 0.73 [0.61;0.80] | 1.000 |
| Neutrophils | 61.8 (9.13) | 61.9 (11.6) | 0.979 |
| NK cells | 10.0 [5.72;13.4] | 7.70 [5.29;17.2] | 0.939 |
| NKT-like cells | 1.24 [0.98;1.96] | 2.34 [1.29;4.02] | < 0.05 |
PPF: progressive pulmonary fibrosis; IQR: interquartile range; FVC: forced vital capacity; FEV1: forced expiratory volume in 1s; DLCO: diffusing capacity for carbon monoxide; ILD: interstitial lung disease; AID-ILD: autoimmune disease-associated interstitial lung disease; HP: hypersensitivity pneumonitis; IIP: idiopathic interstitial pneumonia. Differences in the distribution between groups were assessed using “compareGroups” R package. A Shapiro test was performed for each variable and the appropriate statistic test was selected accordingly to their distribution. Data are expressed as mean±standard deviation (SD), median [and IQR], or n (and %). The significance of bold p value is < 0.05.
 Significant correlations between both, forced vital capacity (FVC) and diffusing capacity for carbon monoxide (DLCO), and the different subtypes of T cell compartment in patients with fibrosing interstitial lung diseases (ILDs); FVC was negatively correlated with CD8+HLA-DR+ T cells (r=−0.53; p=0.001) and central memory CD4 T cells (r=−0.39; p=0.024); FVC and DLCO were negatively related with the percentage of T helper type 1 (Th1) cells (r=−0.59; p=0.0002; and r=−0.43; p=0.014, respectively) and CD4+CD28−HLA-DR+ (r=−0.41; p=0.016; and r=−0.041; p=0.021, respectively). (B) Comparison of % NKT-like cells in non-PPF (blue box) and PPF patients (pink box). (C) Flow cytometry gating scheme to identify NKT-like cells. (D) Correlation between FVC and CD8 T cells in patients with progressive pulmonary fibrosis (PPF) (n=11) vs non-PPF (n=22).")
(A) Significant correlations between both, forced vital capacity (FVC) and diffusing capacity for carbon monoxide (DLCO), and the different subtypes of T cell compartment in patients with fibrosing interstitial lung diseases (ILDs); FVC was negatively correlated with CD8+HLA-DR+ T cells (r=−0.53; p=0.001) and central memory CD4 T cells (r=−0.39; p=0.024); FVC and DLCO were negatively related with the percentage of T helper type 1 (Th1) cells (r=−0.59; p=0.0002; and r=−0.43; p=0.014, respectively) and CD4+CD28−HLA-DR+ (r=−0.41; p=0.016; and r=−0.041; p=0.021, respectively). (B) Comparison of % NKT-like cells in non-PPF (blue box) and PPF patients (pink box). (C) Flow cytometry gating scheme to identify NKT-like cells. (D) Correlation between FVC and CD8 T cells in patients with progressive pulmonary fibrosis (PPF) (n=11) vs non-PPF (n=22).
The main and novel observations of this study are that: (1) in the entire population of f-ILDs, several CD8 and CD4 subpopulations correlate with lung function severity at the time of diagnosis and (2) NKT-like cells levels at diagnosis are associated with PPF.
We found several immunological changes regarding CD8 and CD4 T cell repertoire that correlated with lung function severity at the time of diagnosis in the whole population of f-ILDs (Fig. 1A) including that CD8+HLA-DR+ T cells and CM CD4 T cells negatively correlated with FVC, and CD4+CD28−HLA-DR+ T cells negatively correlated with FVC and DLCO. These observations suggest that an exhausted immune system may have a role in the pathogenesis of f-ILDs because elderly persons also show reduced levels of naïve T cells, a relative increase of memory T lymphocytes, and accumulation of antigen experienced CD4+CD28− T cells.4,5 In fact, in keeping with these observations, previous studies also showed that increased levels of CD4+CD28− T cells (also a marker of immune exhaustion) are associated with poor outcomes in IPF patients.6 Finally, we observed a negative correlation between Th1 cells and both FVC and DLCO at diagnosis, likely indicating a pro-fibrotic role of Th1 cells.7
A major question in this analysis was to investigate if any immune marker determined at diagnosis in a population of f-ILDs at large was associated with PPF. We found that NKT-like cells were increased at baseline in patients who eventually developed PPF as compared to those with non-PPF (Fig. 1B). A previous report showed increased NKT-like cells in bronchoalveolar lavage of f-ILDs.8 Here, we extend these previous observations to circulating blood. NKT-like cells provide protection against pathogens and tumor cells but, of note here, may also be related to cell senescence.9 Additionally, while previous literature suggests a potential influence of smoking on cell populations, the statistical power of the present study was insufficient to convincingly demonstrate the interaction between NK T-like cells decrease and smoking status.
Interestingly, a senescent T-cell phenotype has also been reported in IPF patients.10 Age-related changes in T-cell reservoirs and function includes an expansion of mostly highly differentiated CD8+ T cells, but its role in PPF is unknown. We found that in PPF patients there was a negative correlation between FVC and CD8 T cells (Fig. 1D), which further supports that an accelerated aging of immune response is present in PPF and associates to lung function impairment. This is novel information since, to date, a link establishing interactions between immune cells and functional impairment in PPF had not been established.
Among potential limitations, we acknowledge the limited size of our cohorts. Consequently, undertaken larger studies become crucial to validate these findings. Moreover, it would be valuable to explore the correlation of these findings with the immunological profile in bronchoalveolar lavage or lung biopsies in further studies.
In summary, this study shows that in patients with f-ILDs several immune populations in circulating blood, skewed toward an aged and exhausted immune profile, relate to lung function impairment at diagnosis, and that PPF is associated with an increased cytotoxic immune response. Collectively, these findings highlight biomarkers of potential clinical utility, although we acknowledge that these observations will have to be confirmed in larger cohorts.
FundingThis work was supported by Instituto de Salud Carlos III, FEDER Funds (FIS17/00369, FIS19/01152), PhD4MD program, SEPAR, SOCAP, PFIS predoctoral scholarship (FI19/00054) and the Serra Húnter Professor program.
Conflict of InterestFH-G and JS reports honoraria for lectures, educational events, and support for attending meetings from Roche and Boehringer-Ingelheim, outside the submitted work. XA-R reports honoraria for educational events and support for attending meetings from Boehringer-Ingelheim, outside the submitted work. JS discloses honoraria for lectures, educational events, and support for attending meetings from Astra Zeneca, Gebro, and GSK; consulting fees from Boehringer-Ingelheim and Alofarma, and grants from Roche and Boehringer-Ingelheim, all outside the submitted work. RF reports honoraria for lectures and support for attending meetings from Chiesi and Zambon; grants from GSK, Astra Zeneca, Menarini; consulting fees from GSK, all outside the submitted work. The remaining authors declare no conflicts of interest.
Authors thank all participants in the study for their willingness to contribute to medical research, and the flow cytometry unit of the IDIBAPS for supporting our analysis.
The following are the supplementary data to this article:




www.publicationethics.org.




Archivos de Bronconeumología follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals