





Alpha-1 antitrypsin deficiency (AATD) is a genetic condition resulting in lung and liver disease with a great clinical variability. MicroRNAs have been identified as disease modifiers; therefore miRNA deregulation could play an important role in disease heterogeneity. Members of miR-320 family are involved in regulating of multiple processes including inflammation, and have potential specific binding sites in the 3′UTR region of SERPINA1 gene. In this study we explore the involvement of miR-320c, a member of this family, in this disease.
MethodsFirstly in vitro studies were carried out to demonstrate regulation of SERPINA1 gene by miR-320. Furthermore, the expression of miR-320c was analyzed in the blood of 98 individuals with different AAT serum levels by using quantitative PCR and expression was correlated to clinical parameters of the patients. Finally, HL60 cells were used to analyze induction of miR-320c in inflammatory conditions.
ResultsOverexpression of miR-320 members in human HepG2 cells led to inhibition of SERPINA1 expression. Analysis of miR-320c expression in patient's samples revealed significantly increased expression of miR-320c in individuals with pulmonary disease. Additionally, HL60 cells treated with the pro-inflammatory factor lipopolysaccharide (LPS) showed increase in miR-320c expression, suggesting that miR-320c responds to inflammation.
ConclusionOur findings demonstrate that miR-320c inhibits SERPINA1 expression in a hepatic cell line and its levels in blood are associated with lung disease in a cohort of patients with different AAT serum levels. These results suggest that miR-320c can play a role in AAT regulation and could be a biomarker of inflammatory processes in pulmonary diseases.
La deficiencia de alfa-1 antitripsina (DAAT) es una condición genética que produce enfermedad pulmonar y hepática con una gran variabilidad clínica. Los microARN se han identificado como modificadores de la gravedad de algunas enfermedades y su desregulación podría desempeñar un papel en la heterogeneidad de esta enfermedad. Los miembros de la familia miR-320 regulan múltiples procesos, incluyendo la inflamación, y tienen lugares de unión en la región 3’UTR del gen SERPINA1. En este estudio exploramos la implicación del miR-320c, un miembro de esta familia, en la DAAT.
MétodosPrimero se realizaron estudios in vitro para demostrar la regulación del gen SERPINA1 por parte del miR-320. Además, se analizó la expresión de miR-320c en la sangre de 98 individuos con diferentes niveles de AAT mediante PCR cuantitativa y se correlacionó con los parámetros clínicos. Por último, se utilizaron células HL60 para analizar la inducción de miR-320c en condiciones inflamatorias.
ResultadosLa sobreexpresión del miR-320 en células HepG2 inhibía la expresión del gen SERPINA1. El análisis de expresión de miR-320c en los pacientes reveló una expresión significativamente aumentada en los casos con enfermedad pulmonar. Por otro lado, las células HL60 tratadas con LPS como factor proinflamatorio mostraron un aumento de expresión de miR-320c, lo que sugiere que este miARN responde a procesos inflamatorios.
ConclusiónNuestros resultados demuestran que el miR-320c inhibe la expresión de SERPINA1 en células hepáticas y que sus niveles en sangre están asociados con la presencia de enfermedad pulmonar en pacientes con diferentes niveles de AAT. Esto sugiere que el miR-320c desempeña un papel en la regulación de los niveles de AAT y podría ser un biomarcador de inflamación en enfermedades pulmonares.
Human alpha-1 antitrypsin (AAT) is one of the most important serine proteinase inhibitor primarily synthesized and secreted by hepatocytes.1 The protein exerts its major function in the lungs, where it protects the alveolar tissue from proteolytic damage produced mainly by neutrophil elastase.2 In addition to its role as antiprotease, AAT has important anti-inflammatory and immunomodulatory properties.3
AAT is a polymorphic protein and small variations in its three-dimensional structure affect its susceptibility to polymerization and intracellular retention, and also modify its anti-elastase capacity.4 Some authors have hypothesized that polymers can also cause inflammation by themselves.5,6 Accumulation and degradation of abnormal protein cause reduced AAT levels in the serum characteristic of the AAT deficiency (AATD). AATD is associated with chronic obstructive pulmonary disease (COPD) which mainly manifest as lung emphysema and occasionally, neutrophilic panniculitis and systemic vasculitis.7
The most common and physiologically normal allele is the M allele, while Z (Glu342Lys) and S (Glu264Val) mutations are the most common deficiency variants.8 Homozygous ZZ individuals present with severe AAT deficiency (serum levels usually<57mg/dl).9 Homozygous SS and composite heterozygous (SZ) have a mild and moderate deficiency (range of serum levels measured by nephelometry in our laboratory: 70–105 and 45–80mg/dl, respectively).10,11
AAT deficiency (AATD) is not a disease in itself; instead it is a genetic predisposition to develop lung or liver disease mainly in the presence of environmental factors such as inhaled toxins like tobacco smoke. Even among deficient individuals with similar tobacco exposure, high clinical heterogeneity has been observed, ranging from asymptomatic to severe lung disease.12–14 Then, it is reasonable to hypothesize that additional genetic factors that influence gene expression may also be involved in disease presentation.15
In recent years, the role of different molecules participating in DNA transcription and RNA translation into proteins has become of interest to explain clinical variability in genetic conditions and also for their potential use as biomarkers.16,17 MicroRNAs (miRNAs) are small non-coding RNAs that play a crucial role in post transcriptional regulation. Through the binding to the 3′ untranslated region (UTR), miRNAs inhibit translation of target messenger RNAs (mRNAs).18 miRNAs are involved in many biological processes and abnormal miRNA expression has been associated with various lung diseases such as COPD, asthma and lung cancer.19,17 Because miRNAs are potent disease modifiers,20 miRNA deregulation could play an important role in the phenotypic heterogeneity of AATD. In this context, specific miRNAs could serve as biomarkers in the diagnosis, prognosis and treatment of AATD. Nevertheless, the regulation of SERPINA1 expression by miRNAs has been poorly investigated. According to databases, hundreds of miRNAs are predicted to target SERPINA1 3′UTR, but only one, miR-940, has been experimentally validated so far.21 In addition, few studies have explored the role of miRNAs in the pathogenesis of AATD.19,17,22,23
The miR-320 family of miRNA has been associated with lung disease.24–28 It comprises a group of 6 mature miRNAs encoded by sequences on chromosomes 8 (miR-320a-5p and -3p), 1 (miR-320b), 18 (miR-320c), 13 (miR-320d), X (miR-320d) and 19 (miR-320e) (http://www.mirbase.org/). All of them, except miR-320a-5p, share the seed sequence and are predicted to target SERPINA1 3′UTR (according to TargetScan database).
In the present study we aim to explore whether miR-320c, as a representative member of miR-320 family, could be implicated in AAT regulation and AATD pathogenesis, contributing to explain clinical heterogeneity in the patients.
Materials and methodsGrowth and maintenance of cell linesHuman promyelocytic leukemia cells (HL60), human hepatoma cells (HepG2) and human embryonic kidney cells (HEK293T) were grown in RPMI-1640 (HL60) or DMEM (HepG2 and HEK293T) (Sigma-Aldrich), respectively, both supplemented with 10% FBS (Hyclone), 1% penicillin/streptomycin (Lonza) and 0.5% fungizone (Hyclone). The cells were grown at 37°C in a humidified incubator with 5% CO2.
miR-320 family in silico target predictionTargetScan (http://www.targetscan.org) has been used to investigate if SERPINA1 3′UTR has potential binding sites for miR-320 family. TargetScan predicts miRNA targets based on the conserved complementarity to the seed of the miRNA.29
Exogenous miR-320 expression in HepG2 cellsTo test how members of the miR-320 family might affect SERPINA1 gene expression, the following members, miR-320a-3p, miR-320b, miR-320c, miR-320d and miR-320e were overexpressed in HepG2 cells, and then SERPINA1 gene expression was measured by RT-qPCR. This HepG2 cell line was selected since they express high basal levels of SERPINA1. HepG2 cells were seeded at 5×105 cells per well in 6-well plates and after 24h, cells were transiently transfected with 40nM of miR-320a-3p, miR-320b, miR-320c, miR-320d and miR-320e mimic (Ambion) or a no-miRNA control, using Oligofectamine (Invitrogen). At 48h after transfection, the cells were collected for analysis of SERPINA1 gene expression.
Regulation of SERPINA1 gene by miR-320c through Luciferase reporter assaysIn order to investigate whether miR-320c binds the 3′UTR region of SERPINA1 gene we performed luciferase reporter experiments. The 3′UTR of SERPINA1 was first amplified by PCR from the 10835 HapMan cell line and cloned into the pGL3 Control Vector (Promega), downstream to the firefly luciferase. The cloned PCR products were validated by sequencing. The luciferase experiments were performed in HEK293T cells which were co-transfected with the pGL3 plasmid containing the 3′UTR of SERPINA1 gene and with miR-320c. HEK293T cells were grown to 80% confluence in 96-well plates and transfected using Lipofectamine 2000 reagent (Invitrogen) with 100ng of the firefly luciferase-SERPINA1 3′UTR construct, 7.5ng of the control vector pRL-CMV, and 40nM of miR-320c mimic (Ambion) or a non-targeting control (Ambion). Firefly and Renilla luciferase activities were measured consecutively using the Dual-Glo luciferase Assay System (Promega) 48 and 72h after transfection. Experiments were performed in quadruplicate and normalization was achieved using Renilla luciferase activity.
Gene and miRNA expression analysis by quantitative real time PCR (qPCR) in patientsThis project was designed as an observational case-control study to explore and describe the relationship of miR-320 expression with different chronic lung diseases in a population of individuals with different AAT genotypes. It was approved by the ethics committee of Instituto de Salud Carlos III (Madrid, Spain), as well as local ethics committees when necessary.
A total of 98 individuals with different AAT genotypes, were recruited between 2015 and 2016 from the Pulmonology consultations of 11 hospitals in Spain participating in the Spanish Registry of patients with AATD (REDAAT).30 Written informed consent was obtained in advance from all patients. The clinical characteristics of these cases are summarized in Table 1. For the purpose of the study, patients were stratified based on different parameters. Three groups were established depending on AAT serum levels (<50mg/dl, 50–100mg/dl or >100mg/dl). In addition cases were divided in two groups by the presence of any respiratory disease (yes/no) and those with respiratory disease were sub classified according to their clinical diagnoses (as per their clinical records) into the following categories: Emphysema, Bronchiectasis, Chronic bronchitis and Asthma.
Clinical characteristics of individuals with different AAT genotypes.
| MM healthy | MM COPDa | MS | MZ | SZ | ZZ | Other (rare alleles) | |
|---|---|---|---|---|---|---|---|
| n=12 | n=20 | n=7 | n=10 | n=14 | n=27 | n=8 | |
| Sex | |||||||
| Female | 9 | 4 | 6 | 3 | 5 | 14 | 4 |
| Male | 3 | 16 | 1 | 7 | 9 | 13 | 4 |
| AAT serum levels (mg/dl) | |||||||
| Range | 100–220 | 100–188 | 75–140 | 64–110 | 41–60 | 7–45 | 11–91 |
| AAT deficiency | |||||||
| >100mg/dl | 12 | 20 | 5 | 2 | 0 | 0 | 0 |
| 50–100mg/dl | 0 | 0 | 2 | 8 | 11 | 0 | 4 |
| <50mg/dl | 0 | 0 | 0 | 0 | 3 | 27 | 4 |
| Pulmonary disease | |||||||
| Yes | 0 | 20 | 3 | 6 | 9 | 22 | 7 |
| No | 12 | 0 | 4 | 4 | 5 | 5 | 1 |
| Type of airway disease | |||||||
| Bronchiectasis | 0 | 2 | 1 | 1 | 2 | 11 | 2 |
| Emphysema | 0 | 10 | 1 | 3 | 4 | 18 | 4 |
| Chronic bronchitis | 0 | 11 | 1 | 3 | 2 | 12 | 3 |
| Asthma | 0 | 3 | 0 | 2 | 4 | 2 | 2 |
| FEV1% | |||||||
| Range | 99–123 | 28–77 | 74–113 | 16–128 | 59–128 | 23–118 | 64–98 |
| <50% | 0 | 14 | 0 | 2 | 0 | 7 | 0 |
| FVC/FEV1% | |||||||
| Range | 67–96 | 27–70 | 66–111 | 32–85 | 41–85 | 24–99 | 47–92 |
| <70% | 2 | 19 | 2 | 4 | 3 | 20 | 4 |
| Smoking status | |||||||
| Never | 4 | 1 | 3 | 4 | 6 | 6 | 3 |
| Current | 2 | 9 | 3 | 3 | 3 | 6 | 0 |
| Former | 3 | 10 | 1 | 3 | 5 | 15 | 4 |
| Replacement therapy | |||||||
| Yes | 0 | 0 | 0 | 0 | 1 | 15 | 2 |
| No | 12 | 20 | 7 | 10 | 13 | 12 | 6 |
Regarding their lung disease severity and in order to avoid multiple subclassification in small groups those with respiratory disease were only divided as severe impairment (FEV1<50%) or non-severe (FEV1>50%). FEV1/FVC (<70% predicted) was considered as indicator of airways obstruction. Smoking status was recorded as never, current and former. Administration of AAT replacement therapy was collected only as yes/no. The healthy controls were principally selected among family members of the patients.
The blood samples used for miRNA expression analysis were collected in PaxGene tubes (PreAnalytiX) locally and transported to the main laboratory. RNA isolation was performed following manufacturer recommendations.
To analyze the levels of miR-320c directly in the blood cells of patients, total RNA from blood samples was extracted using miRNeasy Micro kit (Qiagen) that allows purification of total RNA including small RNAs. Quantification of miR-320c expression was performed by qPCR using miRCURY LNA™ Universal RT microRNA PCR system (Exiqon) according to the manufacturer's protocol. Briefly, 100ng of total RNA was reverse transcribed and the resultant cDNA was 10x diluted and amplified with specific LNA primers and SYBR Green Master Mix (Exiqon).
For SERPINA1 expression, 1μg of total RNA was reverse transcribed using Maxima First Strand cDNA synthesis kit (Thermo Scientific). The cDNA was 10× diluted and amplified using fluorescent-labeled probes TaqMan (Roche Universal Probe library, Roche), specific primers and TaqMan Universal PCR Master Mix (Applied Biosystems). Gene-specific primers were as follows: SERPINA1F:tgaggagagcaggaaaggac, SERPINA1R:ctcagccagggagacagg (probe 18); GUSB_F:cgccctgcctatctgtattc, GUSB_R:tccccacagggagtgtgtag (probe 57). All reactions were performed in triplicate and no-template controls were included in each run. For normalization, GUSB was used as reference gene for mRNA expression and SNORA66 for miRNA expression. Relative expression was calculated by using the comparative cycle threshold (ΔΔCt) method.
Pro-inflammatory lipopolysaccharide (LPS) treatment of HL60 cellsSince miR-320c might be related to inflammation in pulmonary disease, we performed in vitro experiments to check how the bacterial lipopolysaccharide LPS, a known factor mimicking many inflammatory effects of cytokines, affects miR-320c expression. We treated HL60 cells with the pro-inflammatory factor LPS and quantitatively analyzed the expression of miR-320c at different time points. HL60 cells were seeded at 5×105 cells per well in 6-well plates and treated with 100nM of LPS from Escherichia coli 0111:B4 (Sigma–Aldrich). At 24, 48 and 72h, the cells were collected for analysis of miRNA expression.
Statistical analysisData analysis was performed with SPSS and GraphPad Prism softwares. Data is represented as mean and standard deviation (SD). A Student's t-test or Mann–Whitney test were used to evaluate differences between two groups, either to compare a group of patients with control healthy group, or between two patient categories separately. p<0.05 was considered statistically significant.
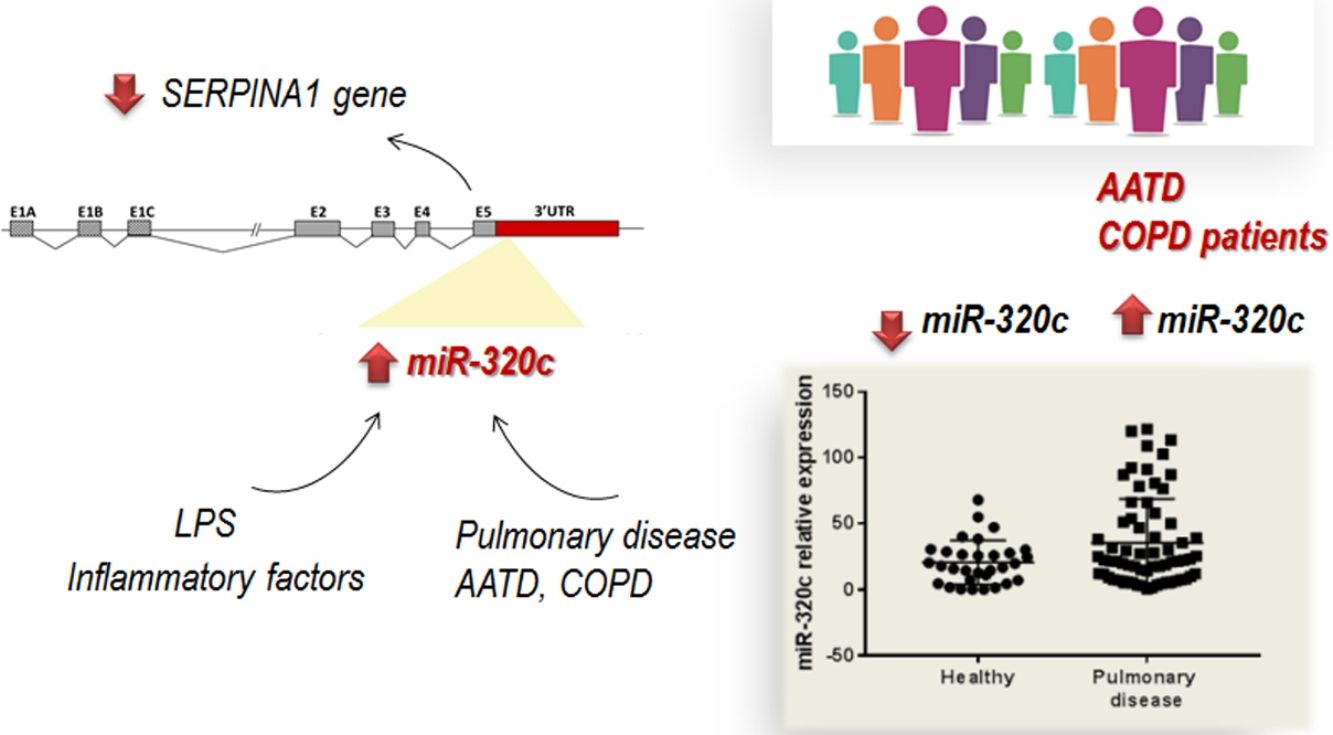
ResultsmiR-320c inhibits SERPINA1 in HepG2 cellsA potential binding site of miR-320 family in the 3′UTR of SERPINA1 was predicted (Fig. 1a). Most of the members of miR-320 family share a similar sequence that is predicted to target SERPINA1 3′UTR at position 256–262. The expression in HepG2 cells of miR-320a-3p, miR-320b, miR-320c, miR-320d but not miR-320e led to an inhibition of SERPINA1 expression (38–68% reduction), as compared to the no-miRNA control (Fig. 1b). Since miR-320c was one of the miRNAs reducing more SERPINA1 expression and was expressed in blood samples, we selected this miRNA for further experiments.
 Schematic diagram showing the predicted binding site on SERPINA1 3′UTR of 5 members of miR-320 family. (B) Relative expression of SERPINA1 after 48h of transfection of miR-320a-3p, miR-320b, miR-320c, miR-320d and miR-320e mimics or no miRNA into HepG2 cells, expressed as percentage. (C) Relative luciferase activity of a reporter vector carrying the SERPINA1 3′UTR downstream of the firefly luciferase gene, expressed as percentage. The vector was co-transfected with miR-320c mimic, non-targeting control or with no miRNA into HEK293T cells. Luciferase activity was analyzed at 48 and 72h. Data represent the mean (± standard deviation, SD) of three independent experiments and are presented relative to mock transfection (no miR). *p<0.05, **p<0.005, ***p<0.001.")
Regulation of SERPINA1 expression by miR-320 family. (A) Schematic diagram showing the predicted binding site on SERPINA1 3′UTR of 5 members of miR-320 family. (B) Relative expression of SERPINA1 after 48h of transfection of miR-320a-3p, miR-320b, miR-320c, miR-320d and miR-320e mimics or no miRNA into HepG2 cells, expressed as percentage. (C) Relative luciferase activity of a reporter vector carrying the SERPINA1 3′UTR downstream of the firefly luciferase gene, expressed as percentage. The vector was co-transfected with miR-320c mimic, non-targeting control or with no miRNA into HEK293T cells. Luciferase activity was analyzed at 48 and 72h. Data represent the mean (± standard deviation, SD) of three independent experiments and are presented relative to mock transfection (no miR). *p<0.05, **p<0.005, ***p<0.001.
Experimental validation of the binding of miR-320c to the 3′UTR, showed marked reduction in luciferase levels (up to 70% at 72h) compared to those cells with no-miRNA transfection (Fig. 1c). These data demonstrate that miR-320c targets SERPINA1 3′UTR and inhibits SERPINA1 in HepG2 cells.
miR-320c is overexpressed in the blood of patients with lung diseaseTo investigate the potential of miR-320c as a disease biomarker in AATD, first we compared expression in 19 healthy non-deficient individuals (AAT>100mg/dl) being 12 MM, and in addition 7 healthy cases with genotypes MS (5 cases) and MZ (2 cases) without symptoms of disease, with 34 subjects with severe AAT deficiency (AAT<50mg/dl) and 25 individuals with intermediate AAT deficiency (AAT between 50 and 100mg/dl). A significant increase of miR-320c in AAT deficient individuals was observed (Fig. 2a). When miR-320c was analyzed in an additional group of 20 COPD non-deficient patients (AAT>100mg/dl), significantly higher levels comparing to healthy non-deficient individuals were observed, similar to the expression levels detected in AAT deficient subjects (Fig. 2a). When all subjects were stratified according to the presence or absence of lung disease, significant higher levels of miR-320c were detected in individuals with disease (Fig. 2b). Stratification of patients according to the type of airway disease revealed significantly higher miR-320c levels in all groups of patients (bronchiectasis, emphysema, chronic bronchitis and asthma) compared with the healthy individuals (Fig. 2c). In addition, when subjects were stratified according to the presence or absence of lung disease and AAT deficiency, the lowest expression of miR-320c was observed in the healthy non-deficient individuals, followed by the healthy deficient subjects and then the non-deficient individuals with pulmonary disease, and the higher levels were detected in the AAT deficient patients with pulmonary disease (Fig. 2d). Although not statistically significant, ZZ patients with pulmonary disease showed a trend to higher expression of miR-320c when compared with asymptomatic ZZ patients (Fig. 2e). Regarding alpha-1-antitrypsin augmentation therapy, the group of ZZ patients with pulmonary disease who were under treatment showed a trend to lower levels of miR-320c compared to those with pulmonary disease and not treated, although the decrease was not statistically significant (Fig. 2f). Finally, we did not find significant differences in miR-320c expression between non-smokers, smokers and former smokers (Fig. 2g, h). These data show that miR-320c is overexpressed in the blood of patients with lung disease.
 34 subjects with AAT serum levels>50mg/dl, 25 subjects with AAT levels between 50 and 100mg/dl, 19 healthy non-deficient (AAT>100mg/dl) subjects and 20 COPD non-deficient (AAT>100mg/dl) patients; (B) 31 healthy individuals and 66 patients with pulmonary disease; (C) 31 healthy individuals, 19 patients with bronchiectasis, 40 with emphysema, 32 with chronic bronchitis and 13 with asthma; (D) 17 healthy subjects with AAT levels>100mg/dl, 14 healthy subjects with AAT levels<100mg/dl, 21 patients with pulmonary disease and AAT levels>100mg/dl and 45 patients with pulmonary disease and AAT levels<100mg/dl; (E) 5 ZZ asymtomatic patients and 21 ZZ patients with pulmonary disease; (F) 5 ZZ asymptomatic patients, 7 ZZ patients with pulmonary disease and not treated and 14 ZZ patients with pulmonary disease under augmentation therapy; (G) 27 never smokers, 26 current smokers and 41 former smokers; (H) 14 healthy never smokers, 13 never smokers with pulmonary disease, 14 healthy ever smokers and 52 ever smokers with pulmonary disease. Single values, mean and standard deviation of miR-320c expression are shown. * p<0.05. PD=pulmonary disease.")
Relative expression of miR-320c in the blood of 98 individuals with different levels of serum AAT. Statistical analysis of miR-320c expression was performed between (A) 34 subjects with AAT serum levels>50mg/dl, 25 subjects with AAT levels between 50 and 100mg/dl, 19 healthy non-deficient (AAT>100mg/dl) subjects and 20 COPD non-deficient (AAT>100mg/dl) patients; (B) 31 healthy individuals and 66 patients with pulmonary disease; (C) 31 healthy individuals, 19 patients with bronchiectasis, 40 with emphysema, 32 with chronic bronchitis and 13 with asthma; (D) 17 healthy subjects with AAT levels>100mg/dl, 14 healthy subjects with AAT levels<100mg/dl, 21 patients with pulmonary disease and AAT levels>100mg/dl and 45 patients with pulmonary disease and AAT levels<100mg/dl; (E) 5 ZZ asymtomatic patients and 21 ZZ patients with pulmonary disease; (F) 5 ZZ asymptomatic patients, 7 ZZ patients with pulmonary disease and not treated and 14 ZZ patients with pulmonary disease under augmentation therapy; (G) 27 never smokers, 26 current smokers and 41 former smokers; (H) 14 healthy never smokers, 13 never smokers with pulmonary disease, 14 healthy ever smokers and 52 ever smokers with pulmonary disease. Single values, mean and standard deviation of miR-320c expression are shown. * p<0.05. PD=pulmonary disease.
Treatment of HL-60 cells with pro-inflammatory factor LPS revealed an effect on miR-320c expression. After 48h of treatment with LPS, a significant increase in the levels of miR-320c was observed in comparison with the non-treated cells (Fig. 3). This increase was even higher at 72h after LPS treatment suggesting a sustained response.
Discussion of three independent experiments. * p<0.05, ** p<0.005.")
The role of specific miRNAs in the pulmonary disease associated with AAT deficiency is largely unknown. Our results regarding miR-320c indicate, first, that this miRNA regulates SERPINA1 expression at least in an hepatic cell line HepG2, and second, that it is overexpressed in the blood of both AATD patients with pulmonary disease and also non-deficient COPD patients, likely because miR-320c might be modulated by inflammatory processes occurring in the disease.
Overexpression experiments in HepG2 cells led us to demonstrate that most of the members of miR-320 family decrease SERPINA1 expression in vitro. Given that miR-320a-3p, miR-320b, miR-320c, miR-320d and miR-320e have the same seed sequence, a similar miRNA-mRNA interaction was expected. The only one that did not inhibit SERPINA1 levels was miR-320e, which presents an A instead of a G in the 3′ region. These results demonstrate that 3′ compensatory sites might play an important role in target specificity,31 and targeting by each specific miRNA must be experimentally confirmed.
Identification of miRNAs regulating AAT expression is of great relevance. It has been suggested that they could serve as a therapeutic strategy by decreasing the expression of mutant isoforms of the protein. Mueller and coworkers32 treated Z-AAT transgenic mice with a dual-function vector encoding for artificial miRNAs derived from miR-155 and for M-AAT resistant to these miRNAs. As a result, misfolded Z-AAT and liver inflammation were reduced and serum M-AAT was increased. Similarly, Eggenschwiler and collaborators33 treated hepatic cells differentiated from patient-specific iPSCs with miR30-styled shRNA and observed a reduction of the polymeric AAT.
Importantly, since miRNAs are easily detectable in blood samples, they hold good promise as diagnostic biomarkers.34–37 We identified significantly increased levels of miR-320c in the blood of patients with pulmonary disease not only associated with the deficiency, as we also found increased levels of this miRNA in COPD patients with normal AAT levels. The increase of miR-320c was observed independently of the type of airway disease (bronchiectasis, emphysema, chronic bronchitis or asthma). These results suggest a potential role of miR-320c as biomarker in lung disease. The analysis of miR-320c in an extended group of patients with different types and degrees of lung disease would be interesting to further confirm these data.
Although we have demonstrated that miR-320c regulates SERPINA1 expression in a hepatic cell line, its role in whole blood could be different being more related to inflammation and lung tissue damage. Expression analysis of miR-320c was performed on whole blood, where different types of cells are present and macrophages and monocytes, the main AAT producers, represent a small percentage of them. The use of purified monocytes and macrophages for future expression analysis would probably give a more clear association between miR-320c expression and AAT serum levels. Nevertheless, to be used as a biomarker of disease, the analysis in whole blood could be more appropriate for a practical application.
It is probable that during pulmonary disease, inflammatory signals lead to increase levels of miR-320c in blood as we have demonstrated in vitro after myeloid cell exposure to LPS. According to our results, expression of miR-320c might be stimulated by inflammatory processes. A number of studies have shown that AAT has significant anti-inflammatory properties and AAT augmentation therapy reduces different inflammatory responses.38–41 Although not significant, a tendency of miR-320c levels to decrease in a small group receiving AAT therapy was observed. The decrease in miR-320c expression in ZZ patients receiving AAT could be caused by these anti-inflammatory properties of AAT. For example, Hassan and collaborators42 demonstrated that AAT decreases miR-199a, miR-598 and miR-320a expression in ZZ monocytes through the inhibition of NFκB. The link between AAT augmentation therapy and miRNA expression alteration must be further investigated.
In conclusion, we have demonstrated that most members of miR-320 family (miR-320a-3p, miR-320b, miR-320c and miR-320d) inhibit SERPINA1 expression in vitro. miR-320c targets SERPINA1 3′UTR and therefore may contribute to decrease levels of AAT. Furthermore miR-320c was found overexpressed in the blood of patients with pulmonary disease, likely as a result of inflammatory signals. Although the link between miR-320c and lung damage should be further investigated, these results suggest that miR-320c could be a biomarker of inflammatory processes in pulmonary diseases.
FundingThis study was financed by grants from the Institute of Health Carlos III (AESI PI14CIII/00070 and PI17CIII/00042) and SEPAR (Sociedad Española de Neumología y Cirugía Torácica), project 92/2014.
Conflicts of interestN. Matamala, G. Gómez-Mariano, S. Martínez, I. Vázquez-Domínguez, Á. Otero-Sobrino, A. Muñoz-Callejas, E. Sánchez, C. Esquinas, A. Bustamante, S. Cadenas, S. Curi, M.T. Martínez, E. Rodríguez, I. Herrero, S. Castillo, J.M. Hernández, I. Blanco, F. Casas, B. Martínez-Delgado: have nothing to disclose.
B. Lara: personal fees from Oracle Fieldwork.
F.J. Michel: personal fees from CSL Behring, non-financial support from GrifolsandCSL Behring.
L. Lazaro: non-financial support from Grifols Movaco SA, and CSL Behring.
M. Torres-Durán: personal fees from Grifols and CSL Behring.
M. Miravitlles: personal fees from AstraZeneca, Boehringer Ingelheim, Chiesi, Cipla, Menarini, Rovi, Bial, Zambon, CSL Behring, Grifols and Novartis, personal fees from AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Bial, Gebro Pharma, CSL Behring, Laboratorios Esteve, Mereo Biopharma, Verona Pharma, TEVA, pH Pharma, Novartis and Grifols, grants from GlaxoSmithKline and Grifols.
We would like to thank to all the REDAAT (Spanish Registry of Alpha-1 Antitrypsin deficiency patients) collaborators.







