

Idiopathic pulmonary fibrosis is defined as chronic fibrosing interstitial pneumonia limited to the lung, with poor prognosis. The incidence has been rising in recent years probably due to improved diagnostic methods and increased life expectancy. In 2013, the SEPAR guidelines for the diagnosis and treatment for idiopathic pulmonary fibrosis were published. Since then, clinical trials and meta-analyses have shown strong scientific evidence for the use of pirfenidone and nintedanib in the treatment of idiopathic pulmonary fibrosis. In 2015, the international consensus of 2011 was updated and new therapeutic recommendations were established, prompting us to update our recommendation for the medical treatment of idiopathic pulmonary fibrosis accordingly. Diagnostic aspects and non-pharmacological treatment will not be discussed as no relevant developments have emerged since the 2013 guidelines.
La fibrosis pulmonar idiopática es una enfermedad intersticial fibrosante limitada al pulmón, con mal pronóstico. Su incidencia ha aumentado en los últimos años, probablemente por la optimización de los métodos diagnósticos y el aumento en la esperanza de vida. En 2013 se publicó la normativa SEPAR sobre el diagnóstico y tratamiento de la fibrosis pulmonar idiopática. Desde entonces, se han publicado los resultados de ensayos clínicos y metaanálisis que han supuesto, con base en la evidencia científica, la introducción de pirfenidona y nintedanib en el tratamiento de la enfermedad. En 2015 se ha actualizado el consenso internacional de 2011, en el que se describen los cambios en las recomendaciones terapéuticas. Debido a ello cabía actualizar el apartado de la normativa sobre el tratamiento farmacológico de la fibrosis pulmonar idiopática. No se tratarán aspectos diagnósticos ni el tratamiento no farmacológico, ya que no se han producido cambios relevantes desde la normativa de 2013.
In 2013, we published the Idiopathic Pulmonary Fibrosis (IPF) Treatment Guidelines.1 Since then, results have emerged from clinical trials with pirfenidone, nintedanib and N-acetylcysteine (NAC)2–4 that have led to changes in the therapeutic management of this disease. In 2015, the 2011 International Consensus on IPF was updated, and new recommendations on the use of these drugs in the treatment of this disease were incorporated.5 These changes have prompted us to update our IPF Treatment Guidelines.
Pharmacological TreatmentIn 2015, recommendations for the treatment of IPF were updated on the basis of new scientific evidence that emerged after the first diagnostic-therapeutic guidelines were published in 2011.5,6 This was a major milestone in the treatment of IPF, as for the first time a Grade A therapeutic recommendation was established for 2 antifibrotic drugs, pirfenidone and nintedanib, on the basis of Level 1 scientific evidence (clinical trials with reproducible results and meta-analyses) according to the GRADE system.7 In 2011, only grade C or D recommendations had been established for treatment (based on expert opinions, retrospective or unvalidated prospective studies or clinical case series). By 2014, the new drugs were providing benefits in terms of a significant reduction in mortality, positioning IPF as one of the few areas in respiratory medicine in which treatment could provide such clinically significant improvements.
Antifibrotic Drugs Showing Therapeutic BenefitPirfenidone (Esbriet®)Pirfenidone is a pleiotropic drug with antifibrotic and antiinflammatory properties, which inhibits the synthesis of profibrinogenic growth factors, such as transforming growth factor-beta (TGF-β) and platelet-derived growth factor (PDGF), fibroblast proliferation, and formation of collagen.8
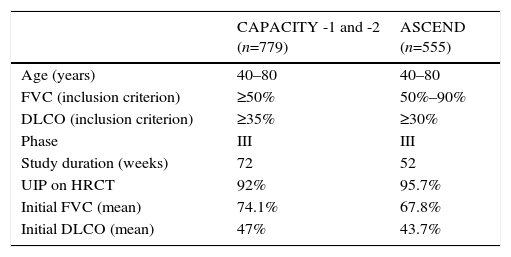
Its clinical efficacy has been evaluated in several double-blind, placebo-controlled randomized clinical trials: 2 Asian studies, 1 phase II and 1 phase III,9,10 and 3 phase III studies in the West (CAPACITY-1, CAPACITY-2, and ASCEND). The 3 Western studies were designed to evaluate the efficacy and safety of pirfenidone in patients with mild-moderate IPF. Table 1 summarizes the main methodological features of the clinical trials. The 2 CAPACITY studies were conducted simultaneously over a 72-week period.8 Meta-analysis of the pooled results of both studies showed less decline in FVC compared to placebo during the 72-week treatment period.8 A Cochrane analysis showed that pirfenidone increased progression-free survival by 30% compared to placebo. European health authorities granted marketing approval to the drug for use in mild-moderate IPF (FVC>50, DLCO>35%), although in the United States, a new study with the same characteristics of the CAPACITY trials was requested to ensure reproducibility of the favorable effects. Thus, the ASCEND study was initiated in 2011,2 in which patients received treatment for 52 weeks. This study again showed a significant delay in functional decline compared to the placebo group, with a relative reduction of 48% in cases of progressive disease or death during the study period. Similarly, patients in the treatment group had significantly longer progression-free survival than patients in the placebo group. Moreover, fewer patients died in the treatment group, and an overall analysis of the data from the CAPACITY-1 and -2 studies, which included 1247 patients, showed a significant reduction in mortality after 1 year of pirfenidone treatment.2
CAPACITY-1 and -2 and ASCEND Clinical Trials: Methodology and Clinical Features of IPF Patients Included.
| CAPACITY -1 and -2 (n=779) | ASCEND (n=555) | |
|---|---|---|
| Age (years) | 40–80 | 40–80 |
| FVC (inclusion criterion) | ≥50% | 50%–90% |
| DLCO (inclusion criterion) | ≥35% | ≥30% |
| Phase | III | III |
| Study duration (weeks) | 72 | 52 |
| UIP on HRCT | 92% | 95.7% |
| Initial FVC (mean) | 74.1% | 67.8% |
| Initial DLCO (mean) | 47% | 43.7% |
DLCO: carbon monoxide diffusing capacity; FVC: forced vital capacity; HRCT: high-resolution computed tomography; UIP: usual interstitial pneumonia.
A meta-analysis evaluating the results from the CAPACITY and ASCEND studies after 1 year of treatment with pirfenidone showed a reduction in the proportion of patients with a decline in FVC≥10% or death (43.8%) and an increase in the proportion of cases with stable FVC (59.3%).11 Benefits in progression-free survival, exercise capacity (measured with the 6-min walk test) and dyspnea were observed. The analysis of these results by patient subgroups showed that the reduction in FVC decline occurs in patients with initial FVC values both above and below 80% predicted value.11
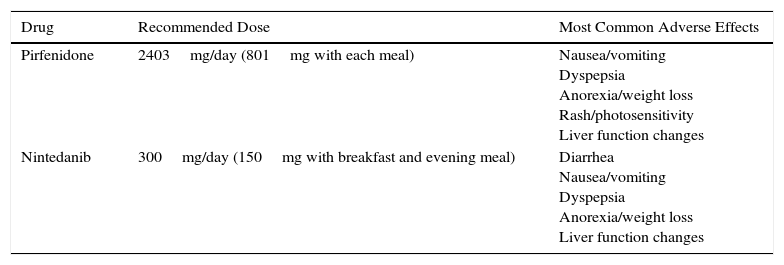
The recommended dose of pirfenidone is 2403mg/day divided in 3 doses to be taken with meals. The maximum dose is reached after 3 weeks of progressive dose escalation: in week 1, one 267mg tablet is administered every 8h, in week 2, two 267mg tablets every 8h and in week 3, three 267mg tablets every 8h. The most significant adverse events are gastrointestinal (nausea, dyspepsia, anorexia, and weight loss), followed by rash, photosensitivity, and to a lesser extent, liver function changes (Table 2). Taking the drug with meals reduces the maximum plasma peak and consequently the risk of adverse effects.12 When gastrointestinal symptoms are significant, the dose may be reduced or even temporarily suspended until symptoms are controlled. When control has been achieved, escalation can be attempted again, until the maximum dose is achieved.12 Prokinetics and proton pump inhibitors (PPI) are useful in the management of gastrointestinal symptoms. However, omprezazole can interact with pirfenidone, so pantoprazole or esomeprazole are recommended if PPIs are required. To minimize the possibility of skin reactions that occur as rash or photosensitivity, patients are advised to avoid direct, prolonged exposure to the sun, to use sun protection cream (PF +50), to wear a hat and sunglasses, and to reduce the areas exposed with appropriate clothing, particularly 1–2h after administration.12
Doses and Main Adverse Effects of Recommended Antifibrotic Drugs.
| Drug | Recommended Dose | Most Common Adverse Effects |
|---|---|---|
| Pirfenidone | 2403mg/day (801mg with each meal) | Nausea/vomiting Dyspepsia Anorexia/weight loss Rash/photosensitivity Liver function changes |
| Nintedanib | 300mg/day (150mg with breakfast and evening meal) | Diarrhea Nausea/vomiting Dyspepsia Anorexia/weight loss Liver function changes |
Accumulated experience after 5 years of use indicates that adverse events and drug withdrawal due to severe adverse effects occur in 3%–4% of cases – an incidence similar to that described in clinical trials.13,14
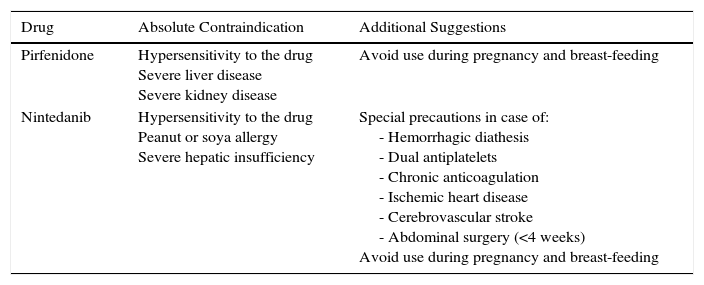
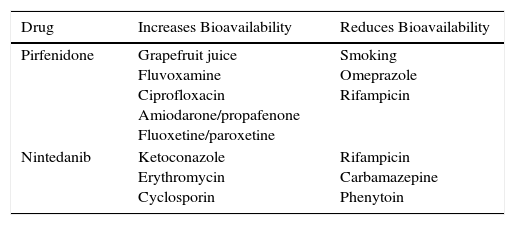
The main contraindications for the use of pirfenidone are drug hypersensitivity, concomitant use of fluvoxamine, and severe liver or kidney disease. Consumption of grapefruit juice should be avoided during pirfenidone administration. No data are available on its use in pregnancy (Table 3). Table 4 shows pirfenidone drug interactions.
Contraindications and Suggestions for the Use of Recommended Antifibrotic Drugs.
| Drug | Absolute Contraindication | Additional Suggestions |
|---|---|---|
| Pirfenidone | Hypersensitivity to the drug Severe liver disease Severe kidney disease | Avoid use during pregnancy and breast-feeding |
| Nintedanib | Hypersensitivity to the drug Peanut or soya allergy Severe hepatic insufficiency | Special precautions in case of: - Hemorrhagic diathesis - Dual antiplatelets - Chronic anticoagulation - Ischemic heart disease - Cerebrovascular stroke - Abdominal surgery (<4 weeks) Avoid use during pregnancy and breast-feeding |
Main Drug Interactions With Recommended Antifibrotics.
| Drug | Increases Bioavailability | Reduces Bioavailability |
|---|---|---|
| Pirfenidone | Grapefruit juice Fluvoxamine Ciprofloxacin Amiodarone/propafenone Fluoxetine/paroxetine | Smoking Omeprazole Rifampicin |
| Nintedanib | Ketoconazole Erythromycin Cyclosporin | Rifampicin Carbamazepine Phenytoin |
Nintedanib is a triple tyrosine kinase receptor inhibitor that has anti-platelet-derived growth factor activity (PDGF), anti-fibroblast growth factor (FGF) activity, and anti-vascular endothelial growth factor (VEGF) activity, all associated with the pathogenic mechanisms of fibrosis. The benefit of nintedanib in IPF is supported by 3 double-blind, placebo-controlled randomized trials.
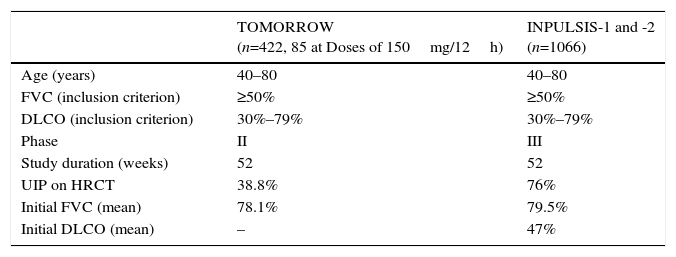
In 2011, the results of a phase II trial (TOMORROW) were published.15 These showed that, compared to placebo, nintedanib at a dose of 150mg/12h significantly delayed lung function decline over a 1-year period and reduced the rate of acute exacerbations. This led to the performance of the phase III studies INPULSIS 1 and 2, the results of which were published in 2014.3Table 5 summarizes the methodological features of these trials.
TOMORROW and INPULSIS Clinical Trials: Methodology and Clinical Features of IPF Patients Included.
| TOMORROW (n=422, 85 at Doses of 150mg/12h) | INPULSIS-1 and -2 (n=1066) | |
|---|---|---|
| Age (years) | 40–80 | 40–80 |
| FVC (inclusion criterion) | ≥50% | ≥50% |
| DLCO (inclusion criterion) | 30%–79% | 30%–79% |
| Phase | II | III |
| Study duration (weeks) | 52 | 52 |
| UIP on HRCT | 38.8% | 76% |
| Initial FVC (mean) | 78.1% | 79.5% |
| Initial DLCO (mean) | – | 47% |
DLCO: carbon monoxide diffusing capacity; FVC: forced vital capacity; HRCT: high-resolution computed tomography; UIP: usual interstitial pneumonia.
In the INPULSIS studies, patients were assigned to receive nintedanib (150mg/12h) or placebo for 52 weeks. The results showed that FVC decline was significantly lower with nintedanib treatment. The INPULSIS-2 trial found a lower percentage of patients with acute exacerbations in the treatment group, and less loss of quality of life. Mortality was lower in the treatment group, but this difference was not statistically significant.3
An analysis of the combined evidence from the TOMORROW and INPULSIS trials showed that nintedanib significantly reduces lung function decline, prolongs time to first acute exacerbation and improves quality of life.16 The subgroup analysis of the data obtained in the 2 INPULSIS studies in 1066 patients found that the delay in disease progression (FVC decline) was independent of age (><65 years), FVC at the time of starting treatment (<>70%, <>90%) and the existence or absence of associated emphysema.3,17,18
The most common adverse effect of nintedanib, occurring in up to 60% of cases, is diarrhea, generally mild-to-moderate in intensity. In only about 4%–5% of patients is it sufficiently severe or recurrent to require definitive withdrawal of the drug. Other less common effects are nausea and vomiting, anorexia, weight loss, and changes in hepatic function (Table 2).
To minimize gastrointestinal side effects, particularly diarrhea, the daily dose can be reduced to 100mg/12h, or administration can even be temporarily suspended until symptoms are controlled. The medication can then be reintroduced and escalated up to the full dose. A binding diet, use of probiotics, and loperamide, combined or not combined with codeine, can help manage the frequency and intensity of diarrhea.
The main contraindications of nintedanib include hypersensitivity to the product, a history of allergic reactions to peanuts or soya, and severe hepatic insufficiency. Caution should be used in patients with hemorrhagic diathesis, dual antiplatelet or chronic anticoagulation, ischemic heart disease, or history of cerebrovascular stroke and recent abdominal surgery (less than 4 weeks). In the INPULSIS studies, patients with cardiovascular risk were excluded, so safety in this population could not be determined (Table 3). Table 4 shows nintedanib drug interactions.
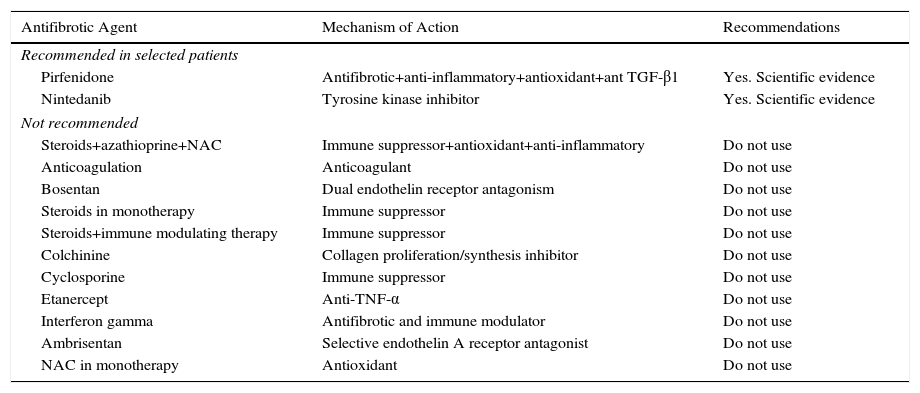
Pirfenidone and Nintedanib in Clinical PracticeWhen a diagnosis of IPF has been established, antifibrotic medication should begin as soon as possible in order to delay functional decline that would occur without treatment.19 In asymptomatic patients with minimal functional or radiological impact, a wait-and-see approach can be taken, after the risks and benefits of such a strategy have been weighed up.20 Taking into account the inclusion criteria of the clinical trials with both drugs (Tables 1 and 2), clinical evidence suggests that pirfenidone and nintedanib can be recommended in IPF patients with mild to moderate functional decline (FVC≥50% and DLCO≥30%)1–3,5,8 (Table 6 and Fig. 1). Patients who present contraindications or intolerance to these drugs should be included in clinical trials. Treatment should be administered for 12 months, and should be continued if the disease improves or stabilizes. If it worsens, each patient should be evaluated to determine if continuing treatment is advisable or if other therapeutic strategies should be adopted. Patients with poor prognosis on diagnosis or with rapidly progressing disease should be referred to lung transplantation units. Patients on transplant waiting lists should continue their antifibrotic treatment, since this has been shown to reduce disease progression and does not increase the incidence of post-transplantation complications.21 In severe disease (FVC>50%, DLCO<30%), the treatment of choice is lung transplantation. Patients who still fail to benefit from drug treatment should be included in clinical trials. The need for palliative care must also be evaluated (Fig. 1).
Recommendations for Antifibrotic Treatment in IPF.
| Antifibrotic Agent | Mechanism of Action | Recommendations |
|---|---|---|
| Recommended in selected patients | ||
| Pirfenidone | Antifibrotic+anti-inflammatory+antioxidant+ant TGF-β1 | Yes. Scientific evidence |
| Nintedanib | Tyrosine kinase inhibitor | Yes. Scientific evidence |
| Not recommended | ||
| Steroids+azathioprine+NAC | Immune suppressor+antioxidant+anti-inflammatory | Do not use |
| Anticoagulation | Anticoagulant | Do not use |
| Bosentan | Dual endothelin receptor antagonism | Do not use |
| Steroids in monotherapy | Immune suppressor | Do not use |
| Steroids+immune modulating therapy | Immune suppressor | Do not use |
| Colchinine | Collagen proliferation/synthesis inhibitor | Do not use |
| Cyclosporine | Immune suppressor | Do not use |
| Etanercept | Anti-TNF-α | Do not use |
| Interferon gamma | Antifibrotic and immune modulator | Do not use |
| Ambrisentan | Selective endothelin A receptor antagonist | Do not use |
| NAC in monotherapy | Antioxidant | Do not use |
NAC: N-acetylcysteine.

A Cochrane meta-analysis conducted in 2003 on the use of corticosteroids in IPF concluded that insufficient clinical evidence was available to justify their use.22 However, until very recently, oral corticosteroids were commonly used in clinical practice, probably because until around 2000–2002, other types of pulmonary fibrosis that respond partially to corticosteroid treatment, such as non-specific interstitial pneumonia, were indistinguishable from IPF. Moreover, the IFIGENIA clinical trial studied the effect of NAC and found that this compound in combination with corticosteroids and azathioprine reduced DLCO (%) compared to the use of the latter 2 without NAC.23 This study led to the use of this triple therapy in IPF. The PANTHER study, initiated in 2011, evaluated the effect of triple treatment compared to placebo and to NAC alone. Less than 1 year after starting the study, the triple therapy arm had to be suspended for safety reasons, as mortality and hospital admission rates were higher than with placebo.24 The placebo and single-agent NAC arms continued until the study was completed. No therapeutic benefit was found for NAC after a 60-week follow-up.4 Thus, there is sufficient evidence to advise against the use of corticosteroids in combination with azathioprine in the treatment of IPF.5 Currently, the use of corticosteroids in IPF is limited to (1) acute disease exacerbations (see the specific section), and (2) cases with difficult-to-manage symptoms, such as refractory cough that does not improve with low dose antitussives (5–10mg/day), occasionally as part of palliative care. In both cases, the clinical evidence for benefit is low.
Antifibrotics in Clinical TrialsClinical trials are currently ongoing with antifibrotics in 2 settings: (1) IPF disease phases that fail to show therapeutic benefit with marketed antifibrotics, including subclinical or incipient disease or more advanced stages (FVC<50%, DLCO<30%). Both marketed antifibrotics and new antifibrotics are being used in single-agent and combination therapy in this setting (www.clinicaltrials.gov). (2) In mild-moderate IPF phases (FVC>50%, DLCO>30%), new proposals for investigational antifibrotics always involve a combination with either of the 2 marketed products. Given the complexity of the multiple pathogenic pathways of the disease, the simultaneous use of different treatment lines may, theoretically, act synergically to generate greater clinical benefit.25,26 The first combination to enter trials was pirfenidone plus NAC after preliminary studies suggested that the combination was potentially synergic.27 Initial safety data with both drugs are positive but no clinical benefit is obtained from combining NAC with pirfenidone.28 Another combination to enter trials is nintedanib+pirfenidone. A recent phase II clinical trial conducted in Asia to evaluate the safety, tolerance and pharmacokinetics of the combination has shown short-term safety, albeit with increased gastrointestinal side effects, particularly nausea and vomiting.29 The phase IV COMBO clinical trial is currently being performed to evaluate 32-week safety. Other combinations of pirfenidone or nintedanib with other new antifibrotics, initiated before either drug received marketing approval, are also ongoing (www.clinicaltrials.gov), including pirfenidone±lebrikizumab (RIFF study), pirfenidone or nintedanib±PRM-151 (PROMEDIOR study), pirfenidone±vismodegib (GB29764 study), pirfenidone±sildenafil.
Treatment of Associated ComorbiditiesGastroesophageal RefluxSome studies have shown the benefit of medical and surgical treatment of gastroesophageal reflux (GER). In a small series of 4 patients with IPF and GER, treating GER with proton pump inhibitors (PPI) or Nissen fundoplication helped stabilize FVC.30 In another retrospective study in 204 IPF patients, Lee et al. found an increase in survival in patients whose GER was treated, primarily with PPIs.31
An analysis of data from 242 patients included in the placebo arms of 3 randomized clinical trials found that patients who had received PPIs or histamine-2 receptor (H2R) antagonists had less FVC decline and fewer acute exacerbations than those who did not receive antacids.32 In the 2011 ATS/ERS/JRS/ALAT guidelines and the treatment update published in 2015, regular use of PPIs in IPF patients was suggested, although the recommendation was conditional, due to the very low level of evidence for the effect estimates.5
In another study, the effect of antacid treatment on disease progression was evaluated in patients assigned to the placebo arm in 3 phase III clinical trials with pirfenidone (CAPACITY and ASCEND). A total of 624 patients were included in the placebo group, of whom only 291 received antacids. No differences were found between treated and untreated patients in disease progression, all-cause mortality, or FVC decline >10%. An insignificant increase was observed in pulmonary infections in patients receiving antacids.33 The effect of treating GER with laparoscopic Nissen fundoplication on disease progression was evaluated in IPF patients with clinical and functional worsening despite antacid therapy. No significant differences were found between FVC decline before surgery and after a 1-year follow-up period.34 Randomized, placebo-controlled studies are required to determine the benefit of antacid treatment in patients with IPF.
Pulmonary EmphysemaNo effective treatment is currently available for combined pulmonary fibrosis and emphysema (CPFE) syndrome,35,36 so it seems logical to base therapeutic decisions on the separate recommendations for emphysema and IPF. Smoking cessation is the main recommendation for the treatment of COPD and IPF. If CFPE is associated with other environmental exposures, these must be avoided. Oxygen therapy is the most appropriate treatment for concomitant hypoxemia and pulmonary hypertension. In order to reduce acute exacerbations and infections, anti-influenza and anti-Streptococcus pneumoniae vaccination is recommended.
For patients with obstructive or mixed functional limitations, inhaled bronchodilators may be prescribed as for COPD. The use of long-acting inhaled corticosteroids has been beneficial in some patients37,38 for improving lung function and reducing the severity of acute episodes. However, the benefit of bronchodilators in CFPE has not been adequately demonstrated. Lung function decline may also differ in CFPE patients with or without airflow limitation, and as such, the efficacy of one or another bronchodilator may differ. Although the evidence is still unclear, inhaled bronchodilators are prescribed in less than 50% of cases,39 but as no definitive treatment is available for IPF, management of comorbidities may be a good therapeutic strategy for improving the course of the disease.39
Although the CAPACITY (pirfenidone) and INPULSIS (nintedanib) studies included patients with CFPE, evidence for the efficacy of the new antifibrotics is limited and insufficient to draw any conclusions in this respect. A recent study found that in most IPF patients, including those with cardiovascular diseases and emphysema, pirfenidone was well tolerated, and a stable disease course could be maintained. However, specific efficacy in CFPE with either pirfenidone or nintedanib has not been proven.11,17
Specific clinical and diagnostic criteria must be established for CPFE, and there is an urgent need to improve understanding of the pathogenesis, pathophysiology and prognostic factors of the CFPE syndrome, to design prospective cohort studies, and to explore new and effective therapeutic strategies.39
Pulmonary HypertensionPH generally appears when IPF is advanced; it is usually mild, but has a negative impact on disease prognosis. Patients with PH and IPF who have chronic hypoxemia must receive home oxygen therapy (strong recommendation, low level of evidence), and lung transplantation must be considered, if this procedure is not contraindicated by age or comorbidities. In 2015, 2 guidelines were published that mention the treatment of IPF-associated PH: the ATS/ERS/JRS/ALAT IPF guidelines,5 in which the recommendation regarding treatment was deferred, and the ESC/ERS guidelines on the diagnosis and treatment of PH,40 in which the use of drugs approved for PH caused by lung disease is not recommended (strong recommendation, low level of evidence).
Dual endothelin receptor antagonists (bosentan, macitentan) have been ineffective in the treatment of PH associated with IPF. The selective antagonist ambrisentan, evaluated in the ARTEMIS-IPF clinical trial,41 is contraindicated due to lack of efficacy and increased disease progression and hospitalization for respiratory complications (strong recommendation, level of evidence low).
The phase II study RISE-IIP (NCT02138825) designed to study the efficacy and safety of riociguat, a soluble guanylate cyclase stimulator of the nitric oxide pathway, in symptomatic PH associated with idiopathic interstitial pneumonias, was suspended after increased mortality and serious adverse events were observed in the riociguat-treated patient group.
Sildenafil, a phosphodiesterase-5 inhibitor, was evaluated in the STEP-IPF study in patients with advanced IPF (DLCO<35%); the subgroup of patients with right ventricular systolic dysfunction showed improvements in the 6-min walk test and quality of life.42 Patients with PH or severe right ventricular dysfunction may be considered for personalized treatment (weak recommendation, low level of evidence). Ongoing clinical trials combining pulmonary vasodilators with antifibrotics will show if this approach is beneficial.
Treatment of ExacerbationsTo date, no randomized clinical trial specifically designed to study the treatment of IPF exacerbations has been conducted. Current recommendations, then, are based on results from the few published case series. The only recommendation given on the basis of these studies is to optimize life support (weak recommendation, very low quality of evidence), although several studies have shown that mechanical ventilation in patients with IPF and respiratory failure is ineffective in the majority of cases. The administration of high doses of corticosteroids should be evaluated in the initial days after the event (methylprednisolone in boluses of 500–1000mg/day for 3 days, followed by high doses of prednisone [0.5mg/kg/day]) (weak recommendation).43 No randomized studies are available to support the addition of a second immunosuppressive drug (azathioprine, cyclophosphamide or cyclosporin). No controlled studies are available to support the use of low-weight molecular heparin in the treatment of exacerbations, unless concomitant venous thromboembolic disease is suspected. The empirical use of wide-spectrum antibiotics is also accepted in clinical practice, given the difficulty in many cases of ruling out an underlying infectious process.43–46 No studies have been conducted to evaluate the effect of the new antifibrotic drugs in the treatment of IPF exacerbations, but data are available that suggest a possible preventive effect.3 Thus, prevention is perhaps the most important intervention in IPF exacerbations: anti-influenza and anti-pneumococcal vaccination must be emphasized, GER symptoms must be treated, and surgical biopsies must be avoided in patients with deteriorated lung function and typical findings of usual interstitial pneumonia on high-resolution computed tomography.
Conflicts of InterestAll the authors have participated in scientific activities organizad by Roche and Bohringher. In addition all the authors are have been membres of the advisory committe for IPF from the same pharmaceuticals.
Please cite this article as: Xaubet A, Molina-Molina M, Acosta O, Bollo E, Castillo D, Fernández-Fabrellas E, et al. Normativa sobre el tratamiento farmacológico de la fibrosis pulmonar idiopática. Arch Bronconeumol. 2017;53:263–269.







