

Chronic Pseudomonas aeruginosa lung infection is the most important risk factor for a poor respiratory outcome in patients with cystic fibrosis, and is associated with higher morbidity and mortality, which is worse the earlier it occurs.1–4
Eradication of the pathogen is essential to avoid chronic colonization, but this can only be achieved in the early stages, so it is essential that the infection is diagnosed early and treated intensively in order to prevent progression to chronicity.5–7
In Spain, newborn screening (NBS) began in 1999 in Catalonia, Castilla-León and the Balearic Islands, and in 2015 it was extended to all communities.8 The diagnosis of cystic fibrosis in the neonatal period allows the early detection of the primary bronchial infection with P. aeruginosa. If intensive and persistent treatment is established as soon as this pathogen is isolated, eradication can be achieved in most cases and chronic colonization can be delayed, improving survival.9–11
The main objective of this study was to describe the prevalence of primary infection, intermittent infection, and chronic infection with P. aeruginosa (according to Leeds criteria12) in patients with cystic fibrosis diagnosed by newborn screening (D-NBS) compared with patients not diagnosed by neonatal screening (ND-NBS) who are being or who have been followed up in our Pediatric Cystic Fibrosis Unit. The secondary objective was to describe the prevalence in both groups in terms of mutations.
This was a retrospective descriptive study that included all patients diagnosed with cystic fibrosis before the age of 18 years from 1985 to 2018. We compared D-NBS with ND-NBS patients (including false negatives on NBS). Patients were sub-classified as no function (NF): patients with 2 mutations with no or minimal CFTR function (group I, II or III mutations), and residual function (RF): patients with a mutation with no or minimal CFTR function and a mutation with residual CFTR function (group IV, V, VI, or VII mutation) or 2 mutations with residual CFTR function. Table 1 summarizes the characteristics of both patient groups.
Characteristics of Patients Diagnosed With Cystic Fibrosis by Newborn Screening and Those Not Diagnosed by Newborn Screening.
| D-NBS | ND-NBS | |
|---|---|---|
| Patients (n) | 56 | 26 (2 false negatives per NBS) |
| Mean age at diagnosis | 1.5 months | 4.38 (0.13–15) |
| Current mean age in years | 10.38 (0.8–18) | 21.5 (5–36)a |
| Sex n (%) | Male: 33 (58.9)Female: 23 (41.1) | Male: 13 (50)Female: 13 (50) |
| Lung transplantation | 0 | 5 |
| Deceased | 0 | 8 |
| NF group n (%) | 36 (64.28) | 19 (73) |
| RF group n (%) | 20 (35.71) | 7 (26.92) |
D-NBS: diagnosed by newborn screening; NBS: newborn screening; ND-NBS: not diagnosed by newborn screening; NF: no function, patients with 2 mutations with no or minimal CFTR function (group I, II or III mutations); RF: residual function, patients with a mutation with no or minimal CFTR function and a mutation with residual CFTR function (group IV, V, VI or VII mutation) or patients with 2 mutations with residual CFTR function.
Patients’ medical records and all cultures of respiratory secretions (from birth to age 18 or January 2018 if they were younger) were reviewed. Cultures were performed monthly and, when positive, weekly until negativization. The treatment used for P. aeruginosa primary bronchial infection was inhaled colistin, tobramycin, or aztreonam (3–6 months) along with oral ciprofloxacin (3 weeks). In chronic infections, continuous inhaled treatment with colistin or 28-day on-off cycles with tobramycin or aztreonam were prescribed. During mild-moderate exacerbations, treatment was oral ciprofloxacin (2–3 weeks) and in severe exacerbations, intravenous (β-lactam combined with an aminoglycoside according to sensitivity testing) therapy was administered.
From a statistical perspective, quantitative variables were described by mean and range, and qualitative variables by relative and absolute frequencies. The χ2 test was used to compare prevalences between groups.
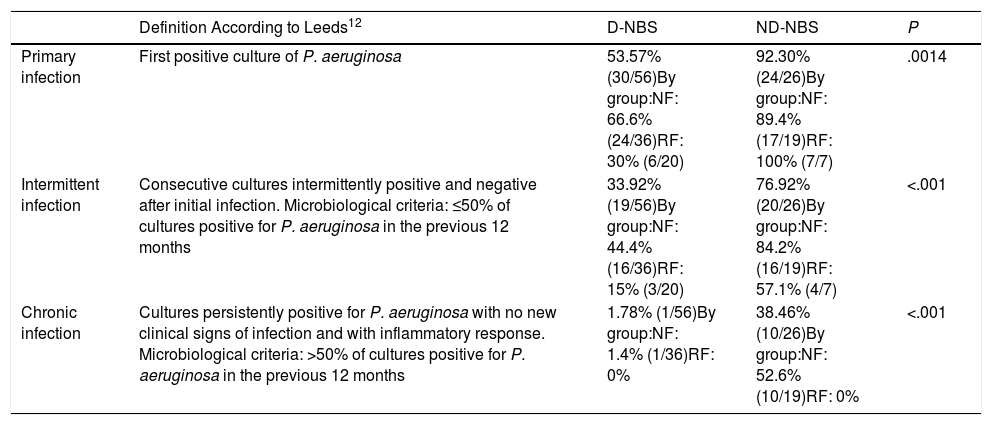
Table 2 shows the prevalences of P. aeruginosa infection in both D-NBS and ND-NBS patients, and in the NF and RF patient subgroups.
Prevalence of Pseudomonas aeruginosa infection in patients diagnosed with cystic fibrosis by newborn screening and those not diagnosed by newborn screening.
| Definition According to Leeds12 | D-NBS | ND-NBS | P | |
|---|---|---|---|---|
| Primary infection | First positive culture of P. aeruginosa | 53.57% (30/56)By group:NF: 66.6% (24/36)RF: 30% (6/20) | 92.30% (24/26)By group:NF: 89.4% (17/19)RF: 100% (7/7) | .0014 |
| Intermittent infection | Consecutive cultures intermittently positive and negative after initial infection. Microbiological criteria: ≤50% of cultures positive for P. aeruginosa in the previous 12 months | 33.92% (19/56)By group:NF: 44.4% (16/36)RF: 15% (3/20) | 76.92% (20/26)By group:NF: 84.2% (16/19)RF: 57.1% (4/7) | <.001 |
| Chronic infection | Cultures persistently positive for P. aeruginosa with no new clinical signs of infection and with inflammatory response. Microbiological criteria: >50% of cultures positive for P. aeruginosa in the previous 12 months | 1.78% (1/56)By group:NF: 1.4% (1/36)RF: 0% | 38.46% (10/26)By group:NF: 52.6% (10/19)RF: 0% | <.001 |
They are also analyzed by patient subgroups: NF group: 2 mutations with no or minimal CFTR function (group I, II or III mutations), RF group: a mutation with no or minimal CFTR function and a mutation with residual CFTR function (group IV, V, VI, or VII mutation) or 2 mutations with residual CFTR function.
D-NBS: diagnosed by newborn screening; NBS: newborn screening; ND-NBS: not diagnosed by newborn screening; NF: no function; RF: residual function.
A comparison of the prevalences of P. aeruginosa infection between D-NBS and ND-NBS patients revealed statistically significant differences in primary infection (P=.0014), intermittent infection (P<.001), and chronic infection (P<.001). This may be due in part to age differences between the 2 groups, since the mean age of the D-NBS patients was 10.38 years and the ND-NBS patients were followed up to the age of 18 years.
Patients with more severe mutations (NF group) diagnosed by neonatal screening had a higher prevalence of primary P. aeruginosa infection than the RF group (P=.018). It should be noted that none of the patients in the RF group (neither D-NBS nor ND-NBS) had chronic P. aeruginosa infection.
Of the D-NBS patients, 53.57% (30/56) had P. aeruginosa infection at some time. Mean age at the time of primary infection was 6 years and the eradication rate was 100%. The median time between first and second infection was 29 months (range: 1.4–96.8). Only 1.78% (1/56) of this group had chronic infection.
Of the ND-NBS patients, 92.30% (24/26) had P. aeruginosa infection at some time. Overall, 19.23% (5/26) already had chronic infection at diagnosis, without eradication. The mean age at diagnosis of these 5 patients was 5.66 years, whereas the mean age at diagnosis for the whole group was 4.38 years. Another 19.23% (5/26) had intermittent infection at diagnosis, but this eventually became chronic. In total, 38.46% (10/26) of this group had chronic infection.
Our results are consistent with other publications, which also show a decrease in the prevalence of chronic P. aeruginosa infection after the introduction of neonatal screening.13–15 In Canada,13 the prevalence of chronic P. aeruginosa infection was 28.4% in D-NBS and 61.8% in ND-NBS patients (P<.001). In the United Kingdom,14 prevalence in children under 15 was 16% in the D-NBS and 20% in the ND-NBS group. The 2018 American Cystic Fibrosis Foundation registry shows that 46.2% of cystic fibrosis patients under the age of 18 had P. aeruginosa infection (17% intermittent and 28.3% chronic) with a median age at primary infection of 5.2 years.15 If we compare the results of these publications with ours, we can see that, although all of them show a decrease in the prevalence of chronic P. aeruginosa infection in D-NBS patients, prevalence in our series is lower. This may be due to very close follow-up, with monthly visits and respiratory secretion cultures, and early and intensive treatment of primary P. aeruginosa infection, and also because there are more patients in the RF group.
In conclusion, we observed a change in the natural history of P. aeruginosa bronchial infection in cystic fibrosis following the implementation of NBS, with a significant decrease (P<.001) in the prevalence of chronic infection with this pathogen in these patients.
FundingThe authors have not received any funding or grants.
Conflict of interestsThe authors state that they have no conflict of interests.
Please cite this article as: Ayats Vidal R, Bosque García M, García González M, Asensio de la Cruz Ó. Infección bronquial por Pseudomonas aeruginosa en los pacientes con fibrosis quística diagnosticados por cribado neonatal. Arch Bronconeumol. 2020;56:532–534.







