






To study the impact of ventilatory management and treatment on the survival of patients with amyotrophic lateral sclerosis (ALS).
MethodRetrospective analysis of 114 consecutive patients admitted to a general hospital, evaluating demographic data, type of presentation, clinical management, treatment with mechanical ventilation and survival. Statistics: descriptive and Kaplan–Meier estimator.
ResultsSixty-four patients presented initial bulbar involvement. Overall mean survival after diagnosis was 28.0 months (95%CI, 21.1–34.8). Seventy patients were referred to the pulmonary specialist (61.4%) and 43 received non-invasive ventilation (NIV) at 12.7 months (median) after diagnosis. Thirty-seven patients continued to receive NIV with no subsequent invasive ventilation. The mean survival of these patients was 23.3 months (95%CI, 16.7–28.8), higher in those without bulbar involvement, although below the range of significance. Survival in the 26 patients receiving programmed NIV was higher than in the 11 patients in whom this was indicated without prior pulmonary assessment (considered following diagnosis, P<.012, and in accordance with the start of ventilation, P<.004). A total of 7 patients were treated invasively; mean survival in this group was 72 months (95%CI, 14.36–129.6), median 49.6±17.5 (95%CI, 15.3–83.8), and despite the difficulties involved in home care, acceptance and tolerance was acceptable.
ConclusionsLong-term mechanical ventilation prolongs survival in ALS. Programmed pulmonary assessment has a positive impact on survival of ALS patients and is key to the multidisciplinary management of this disease.
Conocer el impacto en la supervivencia del manejo y tratamiento ventilatorio de enfermos con esclerosis lateral amiotrófica (ELA).
MétodoAnálisis retrospectivo de 114 pacientes con ingreso consecutivo en un hospital general, evaluando datos demográficos, tipo de presentación, manejo clínico, tratamiento con ventilación mecánica y supervivencia. Estadística: descriptiva y análisis de Kaplan-Meier.
ResultadosSesenta y cuatro pacientes tenían afectación bulbar inicial. La supervivencia media global tras el diagnóstico fue 28,0 meses (IC 95%, 21,1-34,8). Setenta pacientes fueron derivados al neumólogo (61,4%) y 43 recibieron ventilación no invasiva (VMNI) a los 12,7 meses (mediana) del diagnóstico. Se mantuvieron con VMNI sin posterior ventilación invasiva 37 pacientes, cuya supervivencia media fue de 23,3 meses (IC 95%, 16,7-28,8), superior en los no bulbares, aunque en rango no significativo. En 26 en los que la VMNI se indicó de manera programada la supervivencia fue mayor que en 11 en que se indicó sin evaluación neumológica previa (considerando tras el diagnóstico, p < 0,012, y en función del comienzo de la ventilación, p < 0,004). Se trataron en modalidad invasiva 7 pacientes cuya supervivencia fue de 72 meses (IC 95%, 14,36-129,6), mediana de 49,6 ± 17,5 (IC 95%, 15,3-83,8), y pese a las dificultades de la atención en domicilio, la aceptación y la tolerancia fueron aceptables.
ConclusionesLa ventilación mecánica prolonga la supervivencia de la ELA. La evaluación neumológica programada tiene un impacto favorable en la supervivencia de los pacientes con ELA y constituye un elemento esencial en el manejo multidisciplinario de esta enfermedad.
Amyotrophic lateral sclerosis (ALS) is characterized by muscle weakness and progressive paralysis, and is the most common form of motor neuron disease. Incidence is estimated to be around 1–2 cases per 100 000 inhabitants/year: 5%–10% of cases are the familial form, and the majority is the sporadic.1,2 It is caused by the progressive loss of upper motor neurons in the corticospinal tract, bulbar muscles and the spinal cord, leading to weakness and paralysis of the muscles of the upper and lower limbs, face and trunk, including the diaphragm.3 Presentation is heterogeneous, with variable involvement of the muscle groups and differences in prognosis and quality of life, probably reflecting the different mechanisms causing the disease.4 Bulbar and respiratory muscle involvement is characterized by dysarthria, choking, ineffective cough, dyspnea, orthopnea and progressive respiratory failure, generally leading to death within 3–5 years after disease onset.1,5
Treatment options are very limited. One available drug is riluzole, but its action is limited and weak,6 and the only realistic therapeutic measures are palliative. One of the best of these is non-invasive ventilation (NIV). NIV can be used at home, the equipment is user-friendly and controls respiratory failure, making it very well tolerated and accepted by patients and caregivers. As the disease advances, and when bulbar involvement is significant, NIV becomes insufficient and invasive ventilation via tracheotomy must be considered.7
In addition to respiratory failure, patients develop significant changes in their ability to move and communicate, along with severe nutritional disorders,8 giving rise to delicate ethical and social issues regarding intervention and its clinical implications.9 This is a complex situation requiring the coordination of different teams of health workers and caregivers to provide the pulmonary care necessary for the treatment of respiratory failure.10 With aim of defining the role of the pulmonology specialist in the multidisciplinary management of ALS patients, we have reviewed the utility of programmed respiratory care appointments and the impact of mechanical ventilation on the survival of patients attending a general hospital with no dedicated ALS unit.
Materials and MethodsPatient Selection and TreatmentWe performed a retrospective analysis of patients with a diagnosis of ALS consecutively hospitalized in our center between January 1, 2000 and December 31, 2010. This is a third-level general hospital with a catchment area of 550 000 inhabitants. Standard care of patients with suspected ALS in this setting did not include routine referral to a pulmonologist for evaluation of ventilatory requirements, and patients with a diagnosis of ALS (CIE-9:335.20) had to be identified from the discharge reports in the hospital database. Patients with a definitive or probable diagnosis of ALS determined by a neurologist according to the revised El Escorial criteria3 were included. The initial list of 180 patients supplied by the hospital discharge encoders was examined and 66 patients were excluded either because they had neuromuscular processes other than ALS or because they were treated in social welfare palliative care centers not associated with our hospital. Cases with ALS seen only in outpatient clinics and never hospitalized were also excluded. Patients that, at the beginning or during the course of their disease, had obvious difficulty with phonation or swallowing, confirmed by both a neurologist and a pulmonologist, were classified as having bulbar ALS.
After respiratory assessment by the treating pulmonologist, all patients with respiratory failure, according to established criteria,11 were offered a mechanical ventilation device. VS ultra and VS III Resmed ventilators were used in spontaneous timed (ST) and pressure support (PSV) mode with a nasal or oronasal mask. Ventilation began with the patient hospitalized or in the outpatient clinic, starting with low pressures until symptoms were relieved. Oximetry and blood gases were monitored during waking hours and oximetry during sleep. Hospitalized patients were seen by the physiotherapist attached to the hospitalization unit. In the subsequent follow-up, ambulatory oximetry monitoring was used during sleep, parameters were corrected and possible problems due to mask adjustment were checked. Invasive ventilation via tracheostomy (VS II/VS III Resmed ventilator in assist control [ACV] mode and EOLE 3 volumetric respirator) was indicated when patients presented refractory dyspnea, respiratory failure that could not be controlled with NIV, recurrent aspiration, or inability to manage secretions. If expectoration problems and a cough peak flow lower than 270l/min were observed despite assisted cough physiotherapy, a mechanical cough assistant was indicated (Cough Assist, insufflator–exsufflator, MI-E, Emerson) and caregivers were trained in its use. Except in emergency situations, the ventilatory mode was established after conferring with the patient and caregivers. Tolerance was optimized in training sessions. Patients were monitored in face-to-face visits and telephone calls, and were seen at home by a nurse from the equipment supply company. Outpatient physiotherapy was not provided on a regular basis. Date of death was obtained from the death certificate or hospital records, but the immediate cause of death was not considered a study parameter.
Follow-UpThe date limit for follow-up of patients included in the study was May 1, 2011.
Variables AnalyzedData were collected on demographics (age, sex, date of birth and death), clinical features (date of ALS diagnosis, area of bulbar or corticospinal onset and start dates of NIV, invasive ventilation and indication for mechanical cough assistance), pulmonary function at the start of mechanical ventilation (forced vital capacity) and percentage of time with oxygen saturation less than 90% in nocturnal oximetry readings (CT90).
Statistical AnalysisA descriptive analysis was performed of the study parameters with qualitative variables expressed as percentages and quantitative variables as mean or median, standard deviation and range. Survival in each of the subgroups was analyzed according the Kaplan–Meier method. The level of significance for the whole study was set at 0.05. The statistical software package SPSS version 18.0 for Windows was used for data analysis.
ResultsDuring the study period, 114 patients met the inclusion criteria, 57 (50%) men and 57 (50%) women. Patient age at the time of ALS diagnosis was 67.3±10.8 years (mean±standard deviation), with a range of 30–88 years. Disease onset was predominantly bulbar in 64 (56.2%) of cases and non-bulbar in 50 (43.8%).
Most patients, namely 70 cases (61.4%), were evaluated and monitored by a pulmonologist at some time during the course of their disease, while 44 (38.6%) had no pulmonary consultation nor follow-up. For 12 cases (10.5%), their first contact with the pulmonary medicine department occurred when they were admitted for acute respiratory failure. Nine patients (7.9%) had seen a pulmonologist for other reasons before their ALS diagnosis, two cases were being monitored for bilateral phrenic nerve palsy, two for elevated hemidiaphragm, two for sleep apnea–hypopnea, one for tuberculosis sequelae and radiation therapy, one for dyspnea and expectoration of undefined origin, and one for non-obstructive ventilatory changes. Mean time from ALS diagnosis to the first visit to a pulmonologist was 12.17±15.16 months (median 6.34 months).
Table 1 shows the indications for mechanical ventilation. NIV was indicated in 43 of the 114 cases (37.7%). In 11 (25.6%), it was initiated for respiratory failure during hospital admission without prior evaluation by a pulmonologist. Thirty-seven (32.5%) patients continued to receive non-invasive ventilation, while six required subsequent tracheostomy and invasive ventilation. A total of seven patients (6.1%) received invasive ventilation, in one case without having been treated previously with NIV. Mechanical cough assistance was used in 15 cases (13.2%), in one case without simultaneous mechanical ventilation. Pre-ventilation spirometric data were available in 30 of the NIV patients with a percent predicted FVC of 44.1%±20.4%. Nocturnal oximetry before starting NIV was performed in 23 patients; mean CT90 was 18.6%±31.3%.
Type of Ventilatory Support.
| NIV | Invasive ventilation | |
| All cases and ventilatory modes (% of 114) | 43 (37.7) | 7 (6.1) |
| NIV throughout study follow-up | 37 | – |
| Started NIV then switched to invasive ventilation | 6 | 6 |
| Started invasive ventilation directly without prior NIV | – | 1 |
| Bulbar involvement | 26 | 6 |
| Interval between diagnosis and start of ventilation | ||
| Mean±SD (months) | 16.7±17.0 | 28.8±31.0 |
| Median (months) | 12.7 | 20.1 |
NIV: non-invasive ventilation; SD: standard deviation.
By the end of the follow-up period, 106 patients (92.9%) had died. We excluded 2 cases from the survival analysis, since the exact date of death could not be determined. Mean survival of all patients after disease diagnosis was 28.0 months (95% confidence interval [95% CI], 21.1–34.8), and median, 20.0±1.4 months (95% CI, 17.2–22.7).
Mean survival of the 37 patients who received NIV and did not subsequently receive invasive ventilation (20 with predominantly bulbar involvement and 17 non-bulbar) was 23.3 months (95% CI, 16.7–28.8), with a median of 19.7 (95% CI, 14.0–25.4). No significant differences were found in survival between these patients and the 70 cases that did not receive ventilatory support of any kind (38 with bulbar involvement), with a mean survival of 26.7±4.5 months and median 18.9 (95% CI, 15.3–22.6).
Median survival of ALS patients with non-bulbar disease after diagnosis who received NIV was 30.15 months (95% CI, 19.1–41.2), median 24.8±5.4 (95% CI, 14.2–35.3), while in patients with bulbar ALS it was 17.4 months (95% CI, 10.5–24.2), median 13.4±2.4 (95% CI, 8.6–18.11), although the difference was not significant. Survival, when calculated from the start of NIV, was longer in patients without bulbar involvement than in those with, but the difference was not statistically significant (Fig. 1).
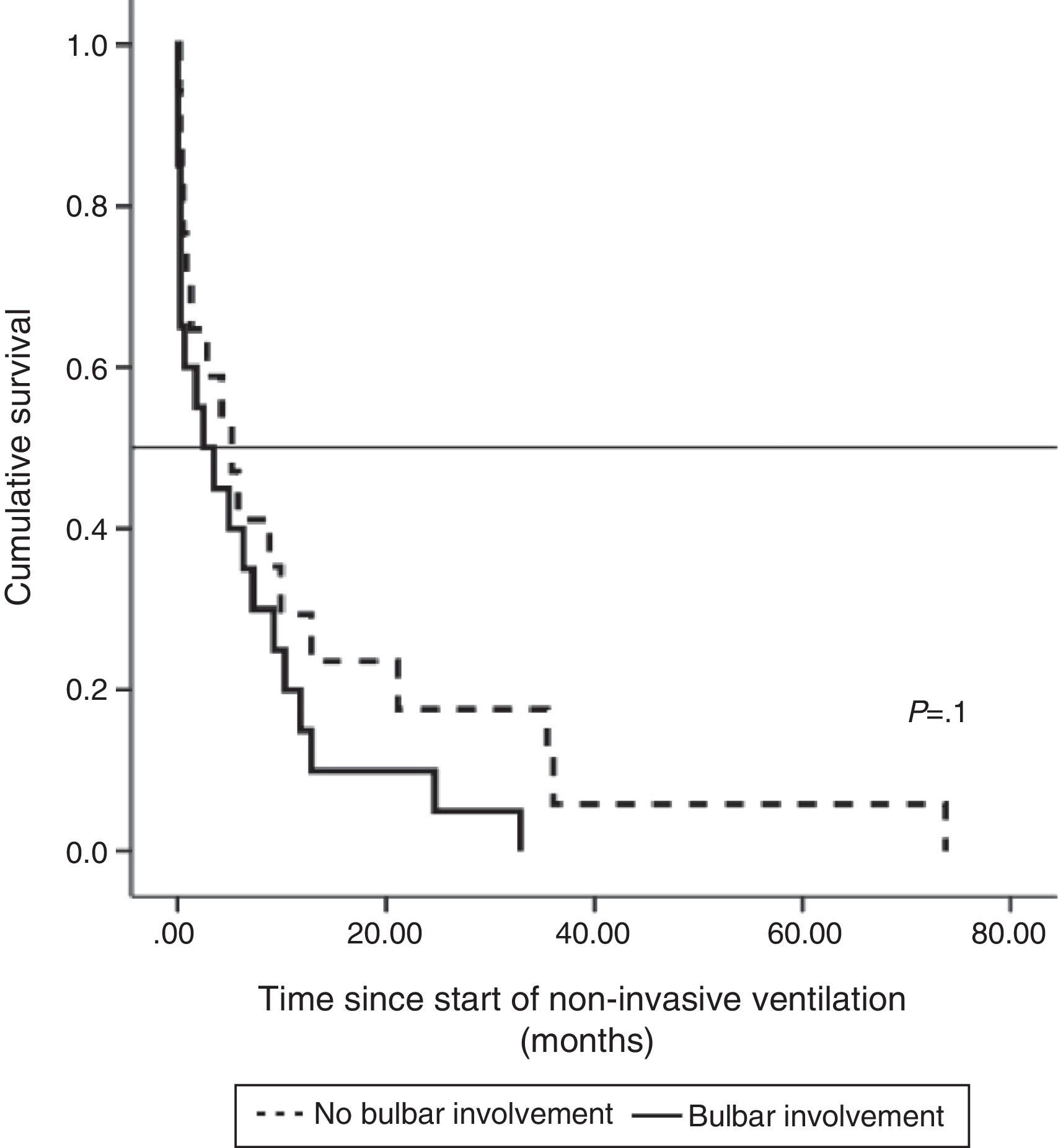
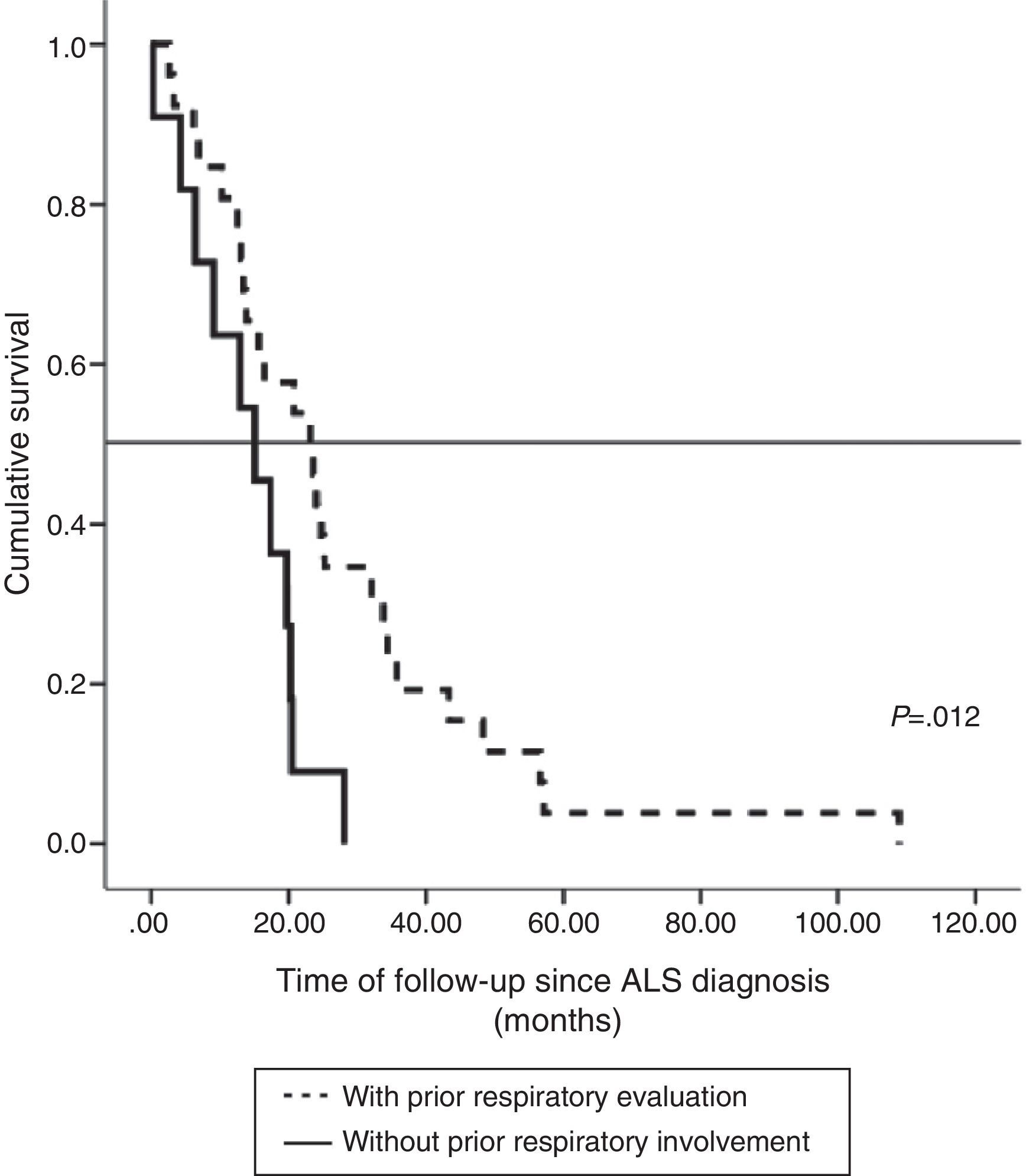
The impact of evaluation by a pulmonologist before ventilatory treatment was analyzed in the 37 patients who received NIV only. In the 26 patients who were prescribed programmed ventilation (13 with bulbar involvement), survival was greater than in the 11 who were prescribed ventilatory support in an emergency situation without prior respiratory evaluation (seven with bulbar involvement). Thus, mean survival, if calculated from the time of ALS diagnosis, was 27.1 months (95% CI, 18.5–35.8) vs 14.0 months (95% CI, 9.1–18.9), P<.012 (Fig. 2), or, from the time of starting ventilation, 12.3 months (95% CI, 5.8–18.7) vs 2.8 months (95% CI, 0.6–5.0), P<.004 (Fig. 3). In patients with prior evaluation by a pulmonologist, time from diagnosis to start of ventilation was 18.7±16.4 months, while in the other group of patients it was 11.5±10.3 months, a difference that was not statistically significant. There were no differences in age, sex and bulbar involvement between the two groups.
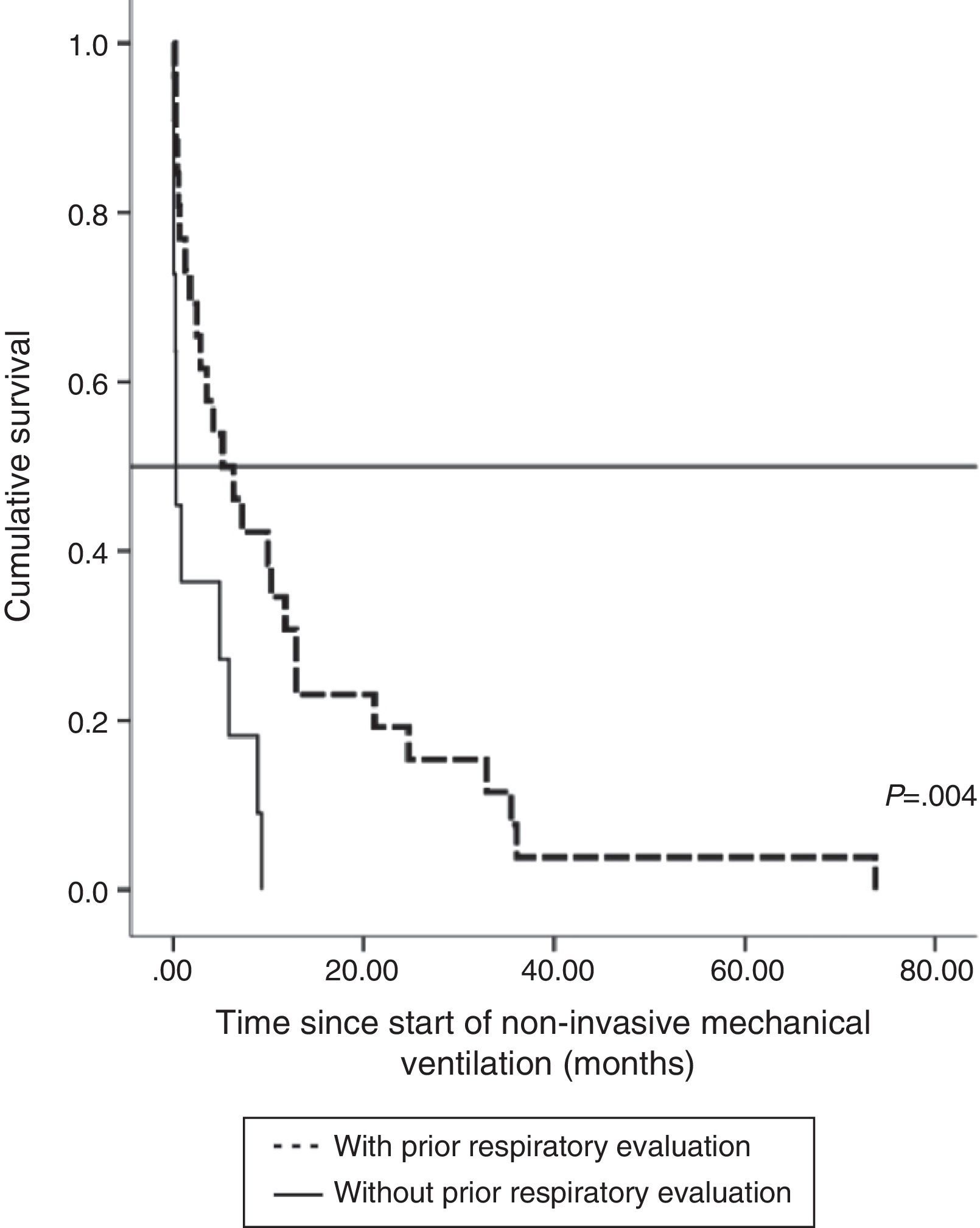
With regard to invasive ventilation, mean survival since diagnosis in the seven patients who received invasive ventilation was 72 months (95% CI, 14.36–129.6), median 49.6±17.5 months (95% CI, 15.3–83.8). This was significantly greater than in the 107 patients who did not receive invasive treatment (P=.01). Of these seven patients, two died within a year after starting invasive ventilation.
DiscussionClinical characteristics of ALS are progressive, severe and incurable paralysis of the voluntary muscles due to destruction of motor neurons.
Patients become dependent for activities of daily living, and when the disease affects the ventilatory muscles, respiratory failure occurs, generally leading to death. No treatment is known to effectively halt the deadly progress of this disease, so when respiratory failure develops, the only recourse is mechanical ventilation.7
In recent years, user-friendly, non-invasive pressure ventilators have become available and have contributed significantly to the management of these patients. These ventilators are not only effective, they are also very well tolerated and are now used both in the hospital and in the home. The efficacy of NIV in ALS is supported by the studies evaluated in two recent systematic reviews, and their impact on quality of life is favorable.12,13 One of the most significant of these studies is that of Bourke et al.,14 due to its randomized and controlled design. These authors randomized 22 patients to NIV and 19 to standard care without mechanical ventilation, and found that in patients without severe bulbar dysfunction, NIV increased survival by 205 days with improved quality of life measured by generic ALS-specific instruments. In patients with severe bulbar involvement, NIV did not increase survival but it did help improve some aspects of quality of life, such as symptoms derived from sleep changes.14 In our study, we found a trend toward improved survival in non-bulbar patients, whose mean survival after NIV was 12.9 months vs 6 months in bulbar patients. This difference was not statistically significant, probably due to the small sample size and the lack of homogeneity between the groups in terms of severity and time since diagnosis. Although survival in patients who only received NIV was not statistically different from that of non-ventilated patients, it is important to bear in mind that in addition to the difference in sample size, these cases had greater respiratory involvement at the time of diagnosis.
Programmed consultation with the pulmonologist, with evaluation of clinical status and respiratory function in patients with suspected or diagnosed ALS, allows the early identification of patients who may respond to NIV.15 The practice in our hospital is probably typical of the standard practice in other similar general hospitals: during the study period we did not have a dedicated ALS unit, and only 43% of patients were referred for a programmed visit with the pulmonologist. Moreover, the 12 most severe cases admitted due to respiratory failure had had no prior respiratory evaluation. It is interesting to note that in nine patients who presented with atypical symptoms, the diagnosis of ALS was made in the pulmonology outpatient clinic, underscoring the fact that this disease can be found in standard respiratory practice.
In our series, referral to the pulmonologist was reflected in health outcomes. Excluding the more severe cases that required invasive ventilation, all patients that were treated with NIV only and were seen for a programmed evaluation before mechanical ventilation was indicated had better survival. This outcome is even better if survival is evaluated from the start of NIV. These results are in line with those of Farrero et al.15
If respiratory deficit is severe and NIV is ineffective, the alternative is invasive ventilation. This decision must only be taken after conferring with the patient and caregivers, and if it is rejected, palliative pharmacological sedation may be offered. These factors explain why only six patients with bulbar involvement went on to invasive ventilation after receiving NIV. Invasive ventilation does not halt disease progression, but it can be used safely in the home, can prolong survival, and is reasonably well tolerated. Sancho et al.16 reported that 78.8% of their patients were still alive one year after starting invasive ventilation. Although our case series is smaller, our experience is similar, with only 2 of the 7 patients dying within 1 year of starting this treatment. Their survival was greater than that of any of our other patients, although the non-invasive ventilation group varied greatly in clinical presentation and severity, and invasive ventilation was refused in some severe cases. In all, survival on invasive ventilation in these cases must be associated with the overall care received by the patient and with the lack of significant complications associated with the tracheostomy, as reported elsewhere.17
In addition to its retrospective nature, this study has other limitations. Patient management was not homogeneous over the long study period, and in the initial years fewer referrals for respiratory evaluation were made. Quality of life was not assessed, sleep studies were not routine, and no real data are available on compliance (although this improved). Patients with admission records were included, but patients who were not hospitalized during the study period were not. The latter are usually patients with less initial respiratory involvement or good family and social welfare support. In contrast, patients who were hospitalized were admitted for severe respiratory failure, adjustment of ventilation parameters, percutaneous endoscopic gastrostomy (PEG) placement, complex tests, or social reasons. Nevertheless, after a 10-year study period, the sample can be said to represent the status quo in our hospital, and in our opinion, is a good reflection of the care practices in a hospital without a dedicated ALS unit.
ALS is a complex disease that is difficult to diagnose; the disease course is complicated, and patients are highly dependent on medical care.3 Protocolized respiratory evaluation and monitoring by pulmonologists prolong survival and improve quality of life. However other needs must also be addressed, such as PEG feeding,18 the use of specialized technology for mobility and communication,19 palliative sedation, and the psychological support and care of patients, caregivers and family.9 This multi-faceted situation can only be effectively dealt with by a multidisciplinary team of professionals willing to share their decisions with patients and caregivers.20
FundingThis study did not receive any financial support.
Conflict of InterestsThe authors state that they do not have any conflict of interests associated with this study.
Please cite this article as: Sanjuán-López P, Valiño-López P, Ricoy-Gabaldón J, Verea-Hernando H. Esclerosis lateral amiotrófica: impacto del seguimiento neumológico y ventilación mecánica en la supervivencia. Experiencia en 114 casos. Arch Bronconeumol. 2014;50:509–513.







