

Alpha-1 antitrypsin deficiency (AATD) is a genetic disorder associated with early onset chronic obstructive pulmonary disease (COPD) and liver disease. It is also a highly under-diagnosed condition. As early diagnosis could prompt specific interventions such as smoking cessation, testing of family members, genetic counseling and use of replacement therapy, screening programs are needed to identify affected patients.
ObjectiveTo estimate the prevalence of severe AATD in COPD patients by routine dried blood spot testing and subsequent genotyping in patients with alpha-1 antitrypsin (AAT) levels below an established threshold.
Materials and methodsCross-sectional study of adult COPD patients attending the Hospital Dr. Antonio Cetrángolo (Buenos Aires, Argentina) between 2009 and 2012. The study consisted of capillary blood collection via finger stick to determine AAT levels, clinical evaluation and lung function tests. Genotype was determined in AAT-deficient patients.
ResultsA total of 1002 patients were evaluated, of whom 785 (78.34%) had normal AAT levels, while low AAT levels were found in 217 (21.66%). Subsequent genotyping of the latter sub-group found: 15 (1.5%, 95% CI 0.75–2.25) patients with a genotype associated with severe AATD, of whom 12 were ZZ (1.2%, 95% CI 0.52–1.87) and 3 SZ (0.3%, 95% CI 0–0.64). The remaining 202 patients were classified as: 29 Z heterozygotes (2.89%, 95% CI 1.86–3.93), 25 S heterozygotes (2.5%, 95% CI 1.53–3.46) and 4 SS (0.4%, 95% CI 0.01–0.79). A definitive diagnosis could not be reached in 144 patients (14.37%, 95% CI 12.2–16.54).
ConclusionThe strategy using an initial serum AAT level obtained by dried blood spot testing and subsequent genotyping was a satisfactory initial approach to a screening program for severe AAT, as a definitive diagnosis was achieved in 87% of patients. However, results were not obtained for logistical reasons in the remaining 13%. This major obstacle may be overcome by the use of dried blood spot phenotyping techniques. We believe this approach for detecting AATD in COPD patients, in compliance with national and international guidelines, is supported by our results.
El déficit de alfa 1 antitripsina (DAAT) es un desorden genético asociado a enfermedad pulmonar obstructiva crónica (EPOC) en edad temprana y enfermedad hepática. A su vez, es una condición altamente subdiagnosticada, lo que haría necesario el desarrollo de programas de cribado para identificar a pacientes afectados, ya que el diagnóstico podría promover intervenciones específicas como cese tabáquico, estudio de familiares, consejo genético y uso de terapia de reemplazo.
ObjetivoEstimar la prevalencia de DAAT grave en pacientes con EPOC mediante la cuantificación de la proteína en sangre seca de forma rutinaria y posterior genotipado de aquellos pacientes con concentraciones por debajo de un umbral establecido.
Materiales y métodosEstudio de corte transversal de pacientes adultos con diagnóstico de EPOC que consultaron al Hospital Dr. Antonio Cetrángolo (Buenos Aires, Argentina) entre 2009 y 2012. La participación en el estudio consistió en la toma de una muestra de sangre por punción capilar del pulpejo del dedo para la determinación de las concentraciones de alfa 1 antitripsina (AAT), evaluación clínica y evaluación de función pulmonar. En los pacientes con déficit, se determinó adicionalmente el genotipo.
ResultadosUn total de 1.002 pacientes fueron evaluados, de los cuales 785 (78,34%) tuvieron un valor normal de AAT, mientras que en 217 (21,66%) se detectó un déficit de concentración de AAT; a este último subgrupo se les realizó genotipado posterior, que arrojó: 15 (1,5%, IC 95% 0,75-2,25) pacientes con genotipo asociado a DAAT grave, de los cuales 12 ZZ (1,2%, IC 95% 0,52-1,87) y 3 SZ (0,3%, IC 95% 0-0,64). Los 202 pacientes restantes se clasificaron como: 29 heterocigotos Z (2,89%, IC 95% 1,86-3,93), 25 heterocigotos S (2,5%, IC 95% 1,53-3,46) y 4 SS (0,4%, IC 95% 0,01-0,79). Por otra parte, en 144 pacientes (14,37%, IC 95% 12,2-16,54) no se llegó a un diagnóstico definitivo.
ConclusiónLa estrategia utilizada con concentración sérica inicial de AAT según la proteína en sangre seca y posterior genotipado resultó adecuada como primera aproximación a un programa de cribado de DAAT grave, ya que se logró el diagnóstico definitivo en un 87% de los pacientes. Sin embargo, no se obtuvieron resultados por razones logísticas en el 13% restante. La implementación de técnicas para fenotipado en proteína en sangre seca permitirá corregir este significativo problema en esta etapa. Creemos que los resultados obtenidos avalarían su aplicación para la detección DAAT en poblaciones de pacientes con EPOC en cumplimiento de las recomendaciones de las guías nacionales e internacionales.
Laurell and Eriksson first described alpha-1 antitrypsin deficiency (AATD) over 50 years ago, after observing the absence of the alpha-1 globulin band in the serum protein electrophoresis patterns of a few patients.1 Its association with chronic obstructive pulmonary disease (COPD) would be discovered later. At present, we know not only the structure of the alpha-1 antitrypsin (AAT) gene, but also the physiopathology of the associated lung and liver disease.
AAT is the most abundant protease inhibitor in human serum, with average concentrations in healthy individuals of 149mg/dL (range: 104–191mg/dL) in our population. Its preferred target is leukocyte elastase, which can digest the elastin in the alveolar walls,2 so much so that its primary function in the lung is to protect the connective tissue from the enzyme released by the neutrophils. The main evidence for this is the development of emphysema in individuals with a deficiency of the protein. In addition to its anti-protease activity, AAT also has immunomodulatory functions, which make it a natural anti-inflammatory molecule.3
The AAT gene is passed on by simple Mendelian inheritance in an autosomal codominant pattern, and is characterized by its extensive polymorphism, with over 100 allelic variants described.4–6 The most common variant in the most severe deficiency is the Z allele. The amount of circulating protein is thought to be directly related with the patient's genotype.
COPD associated with AATD is a potentially fatal disease and a highly under-diagnosed hereditary disorder.7,8 Various series have found that 2%–3% of patients with COPD generally have severe deficiency of the protein. This has led international guidelines to recommend measuring AAT levels in all symptomatic adults with persistent air flow obstruction on spirometry, and in young emphysema patients (aged ≤45 years) or non-smokers.9 However, the deficiency is largely unrecognized, and physicians are reluctant to diagnose it. This is reflected in studies that have found long intervals between the first symptom and definitive diagnosis (a period of 6.3–7.2 years); moreover, 44% of patients report having seen at least 3 physicians before receiving a confirmed diagnosis of AATD.10
Since early diagnosis of the disease could prompt specific interventions (such as smoking cessation, testing of family members, genetic counseling, and replacement therapy indicated in patients with severe deficiency), screening programs have been developed as a strategy to increase detection in the United States.11
Screening, however, is not routinely used in Argentina. Furthermore, as there are no reported data to date in Latin America, we set out to estimate the prevalence of severe AATD in patients diagnosed with COPD by routine dried blood spot (DBS) testing and subsequent genotyping in patients with levels below an established threshold.
Materials and MethodsA cross-sectional study was conducted in adult patients diagnosed with COPD seen in Dr. Antonio Cetrángolo Hospital (Buenos Aires, Argentina) between 2009 and 2012. In accordance with GOLD guidelines, COPD was defined as an FEV1/FVC ratio <70%, based on post-bronchodilator FEV1 lung function tests.12 Spirometry was performed according to ATS/ERS 2005 guidelines,13 using NHANES III reference equations.14
The protocol and informed consent forms were approved by the Dr. Antonio Cetrángolo Hospital ethics committee. All participants agreed to take part in the study. Participation consisted of finger stick capillary blood collection for initial determination of plasma AAT concentrations, a clinical evaluation that included determination of baseline characteristics (demographic, clinical and smoking history) and evaluation of lung function in the lung function laboratory of the aforementioned hospital. Descriptive analysis of these variables was performed; categorical variables were presented as numbers and percentages with their 95% confidence intervals (95% CI), and continuous variables as mean and standard deviation.
Fig. 1 shows the diagnostic algorithm used in the study population. All blood samples were applied to 5 paper discs (DBS, number 903; Schiecher & Schuell; Bioscience Inc., Keene, NH, USA) and allowed to dry at room temperature before being sent to the central laboratory. AAT in DBS was measured by immunonephelometry (Immage®, Beckman Coulter, Inc., CA, USA), in accordance with a previously published method.15 A regression curve was constructed to estimate the AAT concentration in serum from the concentration in the DBS. A cut-off point of 1.80mg/dL in the DBS was validated, corresponding to 100mg/dL of serum AAT.

Genotyping, using real time polymerase chain reaction (PCR) (LightCycler® DNA analyzer, Roche Diagnostics, Mannheim, Germany) with primers for amplification of 2 fragments of 177bp and 229bp, was only performed in patients with AAT levels below the established cut-off value. Specific hybridization probes were used to detect the S (E264V) and Z (E342K) mutations.16 Patients were classified according to the combination of codominant S and Z alleles; SZ and ZZ variants were considered as the genotypes associated with severe deficiency.
Phenotype determination was reserved for patients in whom a discrepancy between AATD deficiency levels and genotype was found. Phenotyping was performed on serum using isoelectric focusing (IEF-Hydrasys®, Sebia, Paris, France). This technique separates the various isoforms of AAT according to their migration on a stable gradient at a fixed pH.
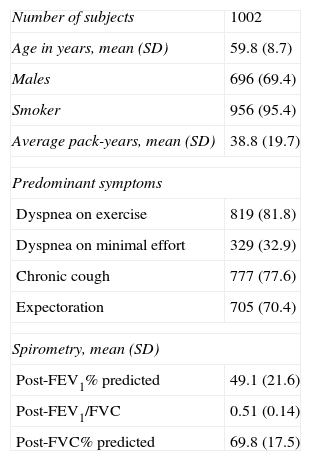
ResultsA total of 1105 eligible patients were identified, of which 103 (9.32%) were excluded. Causes of exclusion were erroneous diagnosis of COPD (54), no sample or insufficient sample (26), and no informed consent (23). Table 1 shows the baseline characteristics of the 1002 patients included.
Baseline characteristics of patients included in the study.
| Number of subjects | 1002 |
| Age in years, mean (SD) | 59.8 (8.7) |
| Males | 696 (69.4) |
| Smoker | 956 (95.4) |
| Average pack-years, mean (SD) | 38.8 (19.7) |
| Predominant symptoms | |
| Dyspnea on exercise | 819 (81.8) |
| Dyspnea on minimal effort | 329 (32.9) |
| Chronic cough | 777 (77.6) |
| Expectoration | 705 (70.4) |
| Spirometry, mean (SD) | |
| Post-FEV1% predicted | 49.1 (21.6) |
| Post-FEV1/FVC | 0.51 (0.14) |
| Post-FVC% predicted | 69.8 (17.5) |
FEV1: forced expiratory volume in the first second; FVC: forced vital capacity; Post-FEV1%: forced expiratory volume in the first second post-bronchodilators (percentage of predicted); SD: standard deviation.
All variables are presented as n (%), unless otherwise indicated.
Based on the AAT levels and established cut-off point, patients were classified as: 785 (78.35%; 95% CI 75.79–80.89) with normal values (>100mg/dL) and 217 (21.65%; 95% CI 19.11–24.21) with AAT deficiency (≤100mg/dL).
Subsequent genotyping of the AATD sub-group detected 15 (1.5%; 95% CI 0.75–2.25) patients with the genotype associated with severe deficiency, of whom 12 were ZZ (1.2%; 95% CI 0.52–1.87) and 3 were SZ (0.3%; 95% CI 0–0.64).
The remaining 202 patients were classified as: 29 Z heterozygotes (2.89%; 95% CI 1.86–3.93), 25 S heterozygotes (2.5%; 95% CI 1.53–3.46) and 4 SS (0.4%; 95% CI 0.01–0.79). In addition, 144 patients (14.37%; 95% CI 12.2–16.54) were lost-to-follow-up, thus preventing a definitive diagnosis; these would have required phenotyping due to discrepant results. However, only 7 patients returned to have a repeat sample taken, obtaining 5 MM and 2 rare M.
DiscussionDue to the lack of data in Latin America, this study is of major importance as a preliminary estimation of the prevalence of AATD in COPD patients in the Argentine population. The estimated prevalence of severe AATD in COPD patients was 1.5% (95% CI 0.75–2.25). Nevertheless, we must acknowledge some limitations in our study: patients with a simple definition of pulmonary obstruction (single lung function test) were included; patients were lost to follow-up due to lack of adherence; and most were from Greater Buenos Aires, with a possible “center-dependent” effect, which prevents us from generalizing the results.
Initial reports correlating AATD deficiency and emphysema had already found a high prevalence of severe deficiencies. For example, in 1969, Lieberman17 studied the trypsin inhibitory capacity of serum specimens from 66 patients admitted to a Veterans hospital for pulmonary emphysema. He identified 10.6% homozygotes and 15.2% heterozygotes. In 1976, Cox et al.18 investigated 163 patients with COPD from the respiratory medicine department of an urban hospital, and found 17.8% of patients with emphysema were PiZ (homozygotes and heterozygotes). In 1986, Lieberman et al.19 found 1.9% PiZZ in 965 individuals with severe COPD. Although they did not give details of the population studied, there is evidence that the patients were in advanced stages of the disease. Based on these and other reports, the World Health Organization (WHO) established prevalence of AATD deficiency in COPD patients at 2%–3%, and recommended AATD screening in COPD patients.20
In a recent study, Wencker et al.21 examined 1060 patients with COPD, asthma and bronchiectasis. They did not find any cases of severe AATD, and the frequency of PiMS and PiMZ detected in this population was similar to that of the normal population. In contrast, a study by Brantly et al.22 reported a 3.2% prevalence of ZZ in 969 patients participating in a free targeted COPD screening program as part of an awareness-raising campaign.
The largest study in COPD patients to date was conducted by Rahaghi et al.23 in 2012. This was a multicenter study carried out in 19 centers in the United States. It included 3457 patients who met ATS/ERS 2005 standardization criteria in their spirometry test, 3152 of whom were considered eligible for the analysis. The reported prevalence of severe AATD was 0.63%, including ZZ homozygotes and SZ heterozygotes. In our study, all patients underwent spirometric tests in the same lung function laboratory, and age groups and male to female ratio was also similar to those in the Rahaghi study.23
There are clearly differences in the reported prevalence of AATD associated with COPD in various studies. This diversity may be due to differing selection criteria, sample sizes, baseline characteristics of the study populations, screening program methodology, and laboratory techniques used.
A key element in the choice of screening strategy in our setting is the overall cost of the program (in terms of the laboratory technique used, sample storage or processing, among others). The DBS method is used worldwide for neonatal screening of metabolic diseases. Its suitability for mass sample analysis has been proven, as it does not necessarily require venous blood extraction, and no special conditions or precautions are needed for sample storage and transport. Both the AAT protein and deoxyribonucleic acid are stable on the paper at room temperature for 1 month, making it possible to send specimens to the laboratory from distant sampling points. Furthermore, analysis is rapid, automated and cost-effective. It is useful for detecting patients with severe AATD, and genotyping can later be performed with the same sample.
Based on the studies consulted,24 our first step in detecting cases was to perform DBS testing to eliminate patients with AAT protein levels above the established cut-off point. Although this cut-off point remains controversial, since it does not ensure that all heterozygous PiMZ or PiMS genotypes are identified, the absence of false negatives for this method enabled us to classify patients with values above this level as “non-deficient”. We believe this to be a reliable method of identifying patients with severe AATD, as the results found in our study are within the ranges reported in the literature.11,22,25,26
More sophisticated studies, such as genotyping or phenotyping, were reserved for patients with concentrations below the selected cut-off point. Genotyping is a rapid technique, but it only detects 2 target mutations: S and Z. Using this approach, therefore, we may have overlooked some PiMZ heterozygotes. Nevertheless, our main aim was to detect patients with severe AATD.
Genotyping and phenotyping should be considered complementary techniques with advantages and disadvantages.27 The rapid genotyping technique for the PiS and PiZ target mutations has been shown to be effective and less laborious. Phenotyping identifies both common and rare alleles in the same gel, but it is time-consuming and considerable experience is needed to interpret the band patterns. Moreover, only one commercial kit and controls is available. In addition, clinically relevant alleles, such as null alleles, may be lost.
Another technique considered to be a confirmatory test is whole gene sequencing. This technique detects any mutation, including de novo variants. However, it is significantly more expensive (approximately 50 times more costly than phenotyping) and is not available in Argentina.
Although Argentine28 and Spanish guidelines29 both propose that phenotyping initially be performed on samples with low AAT concentrations on DBS, we first performed genotyping and then phenotyping, due to the practicality of the first method. If this approach is used in the first stage, an additional sample would not be required and patients would not have to be recalled, which is very difficult at local level. It also reduces the cost of the tests, and yields definitive diagnosis in 87% of cases. However, all patients with absence of the allelic variants S and Z and low AAT levels would have required phenotyping to make a firm diagnosis due to this discrepancy. Most of these patients were not referred to the laboratory due to logistic difficulties, i.e. the relative frequency of AATD could not be estimated in this remaining subgroup (13%). This difficulty could be resolved by implementing techniques for phenotyping in DBS, as a single sample would enable definitive diagnosis.
ConclusionsThe strategy using an initial serum AAT level obtained by DBS testing and subsequent genotyping was satisfactory in this first attempt at a screening program for severe AAT, as it obtained a definitive diagnosis in 87% of patients. However, results were not obtained for logistical reasons in the remaining 13%, a major problem that can be overcome with the implementation of techniques for phenotyping in DBS. We believe that the results obtained support the use of DBS for the detection of AATD in COPD patient populations, in compliance with national and international guidelines that recommend screening.
FundingThis study did not receive any subsidies, contracts, grants or fees.
Conflict of InterestsWe would like to acknowledge the financial assistance provided by Tuteur S.A. to MFA for attending European conferences in 2011, 2012 and 2013; he has also received funding from GlaxoSmithKline for clinical research in COPD.
PBS has received funding from Teva for travel and accommodation at the Argentine Meeting for the definition of national guidelines for the diagnosis and treatment of alpha-1 antitrypsin deficiency, which took place in Iguazú in July 2013, and for the Argentine Congress of Respiratory Medicine in Mendoza, in October 2013.
The other authors declare that they have no conflict of interests.
We would like to thank everyone who contributed to the study, both the authors and those who provided technical and/or statistical help, the directors who provided general support, and especially Dr. Giunta Diego and Dr. Grande Ratti M. Florencia for their help in preparing the manuscript.
Please cite this article as: Sorroche PB, Fernández Acquier M, López Jove O, Giugno E, Pace S, Livellara B, et al. Déficit de alfa 1 antitripsina en pacientes con EPOC: estudio de corte transversal. Arch Bronconeumol. 2015;51:539–543.







