



The most common deficiency alleles for alpha-1-antitrypsin deficiency (AATD) are Pi*S and Pi*S, but there are also other deficiency variants.
This case report describes the first two cases of AATD detected in Spain resulting from the combination of a null Mattawa allele with a normal PI*M, and a rare Mmalton.
Both cases were initially diagnosed as Pi*MM by isoelectric focusing (IEF), but the low serum AAT values led us to suspect the existence of rare deficiency alleles that were undetectable using this technique, and to performing molecular analysis of the gene, which provided the correct diagnosis.
Inconsistencies between serum AAT values and the phenotype should make one suspect the existence of one of these rare alleles.
Los alelos deficitarios más frecuentes son los Pi*S y Pi*Z, pero existen también otras variantes deficientes.
En la presente nota clínica se describen los 2 primeros casos detectados en España de déficit de alfa-1-antitripsina (DAAT), resultante de la combinación de un alelo nulo Mattawa con un normal PI*M y con un raro Mmalton.
Ambos casos fueron inicialmente diagnosticados como Pi*MM por isoelectroenfoque (IEE), pero los valores séricos bajos de AAT hicieron sospechar la existencia de alelos deficientes infrecuentes indetectables por IEE, por lo que se realizó un análisis molecular del gen que proporcionó el diagnóstico correcto.
Las incoherencias entre los valores séricos de AAT y el fenotipo deben hacer sospechar la existencia de uno de estos alelos infrecuentes.
Normal alpha-1-antitrypsin (AAT) alleles, present in 85%–90% of individuals, are called M, and the most common deficient alleles, S and Z (frequencies: 10 and 1.7% of the Spanish population, respectively).1 Patients homozygous for the M, S and Z alleles express around 100, 40 and 15% of serum AAT, respectively.2
Severe AAT deficiency, defined as serum levels below 35% of the mean expected value, is a rare condition, generally associated with PI*ZZ homozygotes and much less frequently with combinations of Z, S, rare and null alleles. However, in recent years, the Spanish AAT Deficiency Registry (REDAAT) laboratory has detected rare and null alleles in 1.6% of cases3 (a rate comparable with that found in Italy, Switzerland, Germany and the USA),4,5 mostly Mmalton, but also Mheerlen, Nullclayton, Nullbellingham, Mvhebron and Ybarcelona, among others.6–8
Case reportCase 1The first patient was a 47-year-old woman, non-smoker, with essential hypertension, recurrent bronchitis since age 25, and exertional dyspnea in the last 3 years.
Her spirometry was normal (FVC 96%, FEV1 105%, FEV1/FVC 84), but her carbon monoxide diffusing capacity (DLCO) and transfer coefficient (KCO) were 68 and 61% of their respective theoretical values. Chest computed tomography (CT) showed an accentuated bronchial pattern with no signs of bronchiectasis or emphysema. Liver function tests were normal. The serum AAT concentration was 73.7mg/dL (reference values: 103–200), consistent with partial AAT deficiency, but the phenotype labeled as PI*MM did not agree with the AAT concentrations. It was therefore decided to perform gene sequencing of the AAT in peripheral blood, in which a PI*Nullmattawa allele was detected. This mutation is characterized by the insertion of a nucleotide within the coding region of exon 5, and results in a frameshift that creates a premature stop signal at position 376 (p.Leu353PhefsX24). This finally translates into a very unstable truncated protein that is degraded inside the hepatocyte and is undetectable in serum9 (Fig. 1).
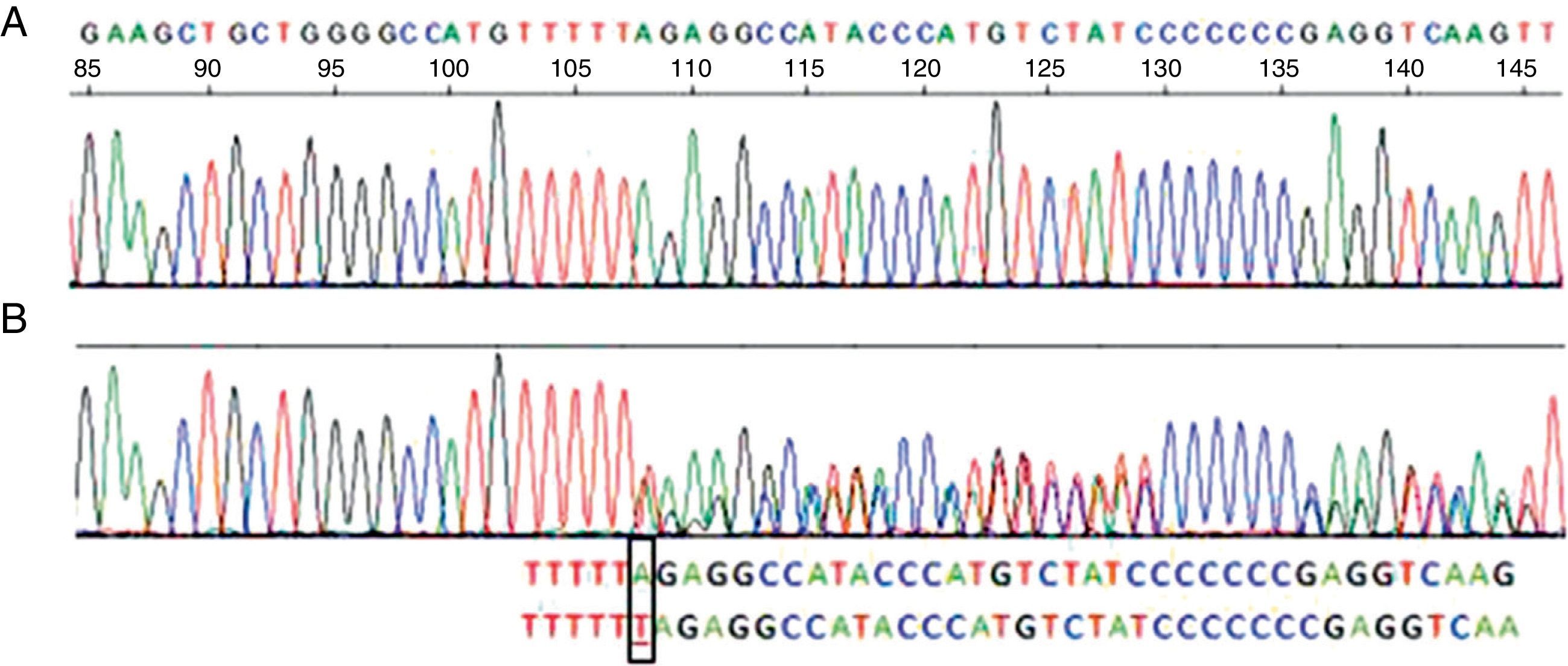
Sequence corresponding to exon 5 SERPINA1. (A) Normal sequence. (B) Sequence corresponding to the patient, which shows insertion of a thymine (T) instead of an adenine (A) at codon 376 in exon 5 of heterozygosity for the PI-Mattawa allele. The SERPINA1 gene coding sequence (exons 2–5) was analyzed using previously described primers for exons 3–5 and 5′ACGTGGTGTCAATCCCTGATCACTG3′ Ex2F primers and ex2R 5′TATGGGAACAGCTGG3′ for exon 2, with reference to the comparative SERPINA1_Transcript_ENST00000440909.
The second allele of the patient contained a normal variant in exon 5, consisting of a substitution of a cytosine (C) for an adenine (A) in nucleotide 1200 of the cDNA, generating the replacement of a glutamic acid (Glu) by an aspartate (Asp) in codon 400 (c. 1200A>C/p.Glu400Asp), which corresponded to an M3 allele. Therefore, this is a PI*M3Nullmattawa heterozygous genotype, with moderately reduced serum AAT levels.
Case 2Another carrier of the Nullmattawa allele was identified from the REDAAT database.10
The patient was a 67-year-old woman with a history of pleuro-pulmonary tuberculosis, with documented bacteriological cure. She suffered frequent respiratory infections. Functional parameters were consistent with severe airway obstruction (FEV1: 620ml and FVC: 1280ml, 22% and 39% of the theoretical values, respectively). High resolution computed tomography (CT) showed diffuse centrilobular and paraseptal emphysema and diffuse cylindrical bronchiectasis. No result indicating liver involvement was detected at any time.
Her serum AAT concentrations were 43mg/dL. IEF suggested a PI*MM phenotype, but as it did not correspond with the serum AAT concentrations, a genetic study was performed. This detected a PI*Mmalton allele (deletion of residue 52, p.Phe52del)11 and a PI*Nullmattawa allele, the combination of which explained the patient's low serum AAT concentrations.
DiscussionThe exceptional nature of the null alleles (estimated prevalence 100–200 times lower than the Z alleles) precludes us from having precise knowledge of their clinical impact and real incidence. Nevertheless, it is important to take them into account, as they can create diagnostic confusion if genomic studies are not performed.
The Mmalton allele (also known as Mcagliari and Mnichinan) was first described in 1987 and, like the Z gene, produces a poorly folded protein. Around 80%–90% of this variant polymerizes in the hepatocyte without being secreted into the blood, where it expresses levels less than 15% when in homozygosity. It is associated with a high risk of pulmonary emphysema and liver disease.11,12
The Mattawa allele (and in general any null allele) codes for a truncated protein, with major conformational changes, which is degraded intracellularly without having the opportunity to aggregate, and hence expresses undetectable serum AAT concentrations. This means that homozygotes have a very high risk of emphysema, but not of liver disease.
In conclusion, in our patients, the discrepancy between the IEF phenotype and the serum AAT concentrations led us to suspect the existence of rare alleles. Molecular analysis of the gene finally provided the correct diagnosis, as stated in the recommendations.2
Conflict of InterestThe authors declare no conflict of interest.
The authors thank Dr. Ignacio Blanco for his important contribution to the writing of this article.
Please cite this article as: Lara B, Martínez-Delgado B, Torres ML, Marín-Arguedas S, Bustamante A, Miravitlles M. Déficit de alfa-1-antitripsina asociado a la variante Matawa. Arch Bronconeumol. 2013;49:548–550.







