
Este estudio ha recibido las siguientes subvenciones: SAF2004-00684, V-2003-RED-C11-FO, BAE 97/5490 y ABEMAR.
Introducción
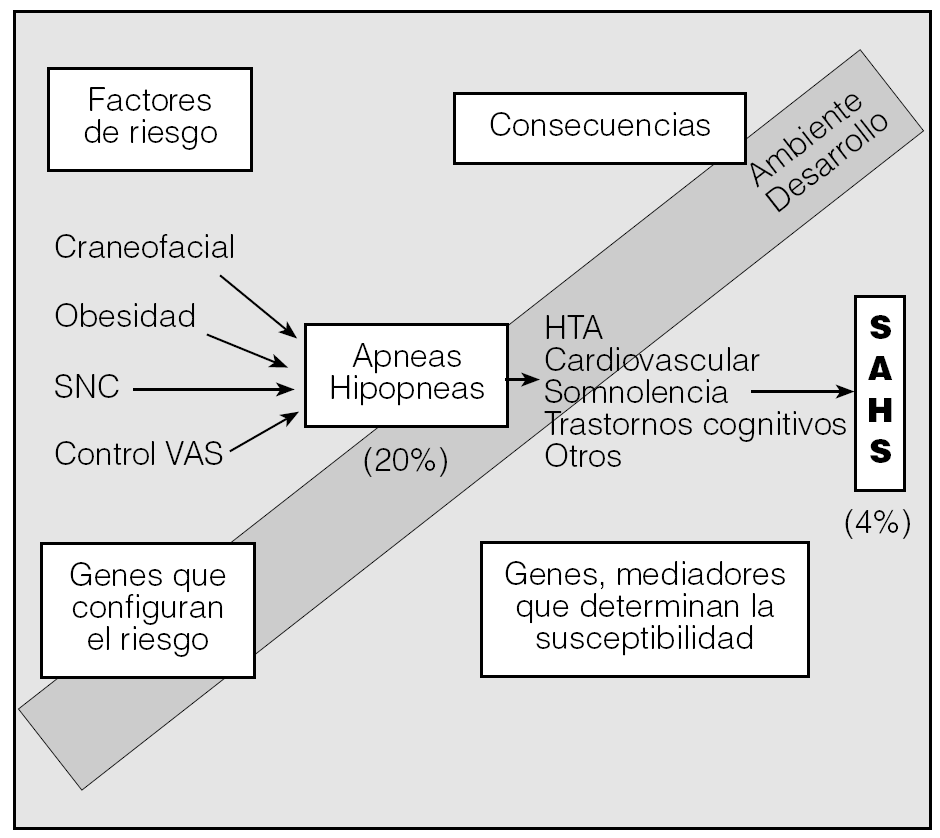
El síndrome de apneas e hipopneas durante el sueño (SAHS) se define como la aparición de episodios de cese total o parcial del flujo aéreo en la boca y la nariz con síntomas secundarios. Hasta la fecha, los estudios se han centrado en aspectos de diagnóstico o tratamiento. Sin embargo, en la actualidad es ineludible abordar nuevos retos como la inflamación, el SAHS y su implicación vascular o el estudio de la vía aérea superior (VAS), que es el nudo etiológico. Además, hay que considerar que existen en el SAHS unos genes que determinan la presencia de una serie de factores de riesgo, lo que explicaría que un 15-20% de la población tenga un índice de apneas-hipopneas superior a 10. El hecho de que la enfermedad acontezca en un 4-6% podría explicarse por una serie de genes que variarían la expresión de mediadores responsables de los síntomas (fig. 1). Por otra parte, que la obstrucción de la VAS se produzca por la noche y no durante el día induce a pensar en la ausencia de unos mecanismos durante la noche para mantener abierta la VAS. En esta revisión se aborda la inflamación en generar el SAHS, la inflamación y la enfermedad vascular y además se profundiza en aspectos de la VAS.

Fig. 1. Esquema que muestra 2 conceptos. Por un lado, una serie de factores de riesgo determinan la presencia o no de apneas. Por otro, las apneas van a produir clínica en función de una serie de factores que expresarán o no una serie de mediadores. HTA: hipertensión arterial; SNC: sistema nervioso central; SAHS: síndrome de apneas-hipopneas durante el sueño; VAS: vía aérea superior .
La inflamación
La inflamación es una reacción inespecífica del tejido conjuntivo vascular que actúa como respuesta protectora del organismo frente a diversas noxas1-3. Consta de 3 eslabones: a) al inicio, cambios del flujo vascular; b) adhesión de células al endotelio y su paso a los tejidos, y c) en los tejidos los linfocitos se activan y son responsables de la inmunidad celular. Los 2 primeros puntos corresponderían a la respuesta aguda, que comporta edema y migración de leucocitos, y el tercero, a la respuesta tardía, que opera a través de macrófagos y linfocitos. En ocasiones se produce con fibrosis y/o necrosis del tejido. Los 3 eslabones interaccionan constantemente.
Eslabón 1
Frente a una noxa que ha franqueado la barrera epitelial (primera barrera), los elementos correspondientes a la inmunidad natural, que existe perennemente (neutrófilos, macrófagos, linfocitos citolíticos naturales, el complemento, la proteína C reactiva y las citocinas --factor de necrosis tumoral, interleucina (IL) 12 e interferón--) se activan liberando sustancias que dan lugar a los procesos básicos de este eslabón: edema, vasodilatación y la llegada de neutrófilos. Estos cambios están mediados por aminas vasoactivas (histamina, serotonina y derivados de la agregación plaquetaria), proteasas plasmáticas (cininas, coagulación, complemento) y los lípidos bioactivos (prostaglandinas y leucotrienos), que, además de colaborar en la reacción vascular inicial, la amplifican e intervienen en casi todo el proceso inflamatorio. A continuación se detallan las funciones de estos mediadores.
El sistema cinina (calicreína y bradicinina) actúa en la superficie de los tejidos. Tiene una función similar a las aminas vasoactivas y aumenta la permeabilidad. El sistema de la coagulación potencia las cininas y el complemento. El complemento proteínas plasmáticas que favorecen la fagocitosis, la destrucción de noxas y la liberación de sustancias por parte de los mastocitos. Estos últimos liberan histamina, lípidos bioactivos (véase más adelante) y citocinas al desgranularse. Tras una segunda exposición a un determinado antígeno mediante la intervención de inmunoglobulina E y eosinófilos, los mastocitos se desgranulan (respuesta alérgica aguda). Los eosinófilos amplifican y perpetúan la reacción (respuesta tardía). Los lípidos bioactivos se liberan del ácido araquidónico de la pared celular. Se pueden activar 2 vías enzimaticas, la de las ciclooxigenasas 1 y 2, que producen prostaglandinas y tromboxano, y la vía lipogenasa, que origina leucotrienos. Las prostaglandinas intervienen sobre aspectos vasoactivos. Los leucotrienos contraen el músculo liso bronquial, favorecen la quimiotaxis y aumentan la permealidad vascular. Otro lípido bioactivo importante es el factor activador de plaquetas, derivado de la membrana celular de las plaquetas, leucocitos y células endoteliales. Interviene de forma decisiva en multitud de eslabones de la respuesta inflamatoria. Provoca agregación plaquetaria, leucocitosis, contracción del músculo liso y aumento de la permeabilidad capilar. Favorece la quimiotaxis y la génesis de radicales libres.
Eslabón 2
Una vez activados los elementos que corresponden a la respuesta inicial, se produce el rodamiento de diversas estirpes celulares en el endotelio y se generan diversas moléculas que favorecen su adhesión y migración a los tejidos. Son las moléculas de adhesión. Las selectinas favorecen la adhesión de neutrófilos, las moléculas de adhesión vascular favorecen a los monocitos, que se van a convertir en macrófagos, y la molécula de adhesión a las células vasculares favorece a los linfocitos. La migración se ve facilitada por una mayor separación entre las células endoteliales a través de la histamina, leucotrienos, cambios en el citosqueleto y por lesiones directas. Asimismo, el endotelio genera mediadores vasoactivos (óxido nítrico, prostaciclina, endotelina 1 y tromboxano) que regulan el tono vascular.
Eslabón 3
En los tejidos los neutrófilos y macrófagos emigrados tienen una acción bactericida mediante la liberación de diversas sustancias (véase más adelante) y además activan los linfocitos T que desarrollan su actividad citolítica (CD8) y activadora (CD4). Los macrófagos, a través de citocinas, entre otros elementos, favorecen la acción de las moléculas de adhesión y, en consecuencia, una mayor incorporación de macrófagos y neutrófilos. Los linfocitos CD8 atacan la membrana de la noxa y causan lisis osmótica. Los linfocitos CD4, a través de la producción de factor de necrosis tumoral, interferón y otras citocinas, dan lugar a un reclutamiento de leucocitos, la activación de fagocitos y la consiguiente destrucción de la noxa. Toda esta reacción celular persigue destruir todo tipo de noxa, aunque en ocasiones se lesionan los tejidos sanos adyacentes. Los mediadores implicados son:
Las citocinas son los mensajeros intracelulares. Su actividad es amplísima; tienen función hematopoyética (IL-3 e IL-13), intervienen en el proceso inflamatorio inicial (factor de necrosis tumoral, IL-1, IL-6, IL-12 e interferón) y también en el tardío (IL-2, IL-4 e interferón). Respecto a la inflamación se dividen en proinflamatorias (p. ej., IL-6) y antiinflamatorias (p. ej., IL-10). La mieloperoxidasa, los gránulos azurófilos, la lactoferritina y los radicales libres son productos liberados por los neutrófilos y macrófagos para su actividad lítica. Los radicales libres tienen una actividad muy amplia. Si se vierten al medio extracelular, dañan las estructuras vecinas. Los antioxidantes controlan su génesis y liberación. Los radicales libres son un elemento fundamental en la génesis de las lesiones cardiovasculares inducidas por el SAHS. La liberación de radicales libres junto a la presencia de otras sustancias como lípidos y células espumosas podría dar lugar a inflamación de los vasos, fibrosis tisular, lesiones vasculares y arteriosclerosis.
El proceso inflamatorio puede ser incluso nocivo cuando es exagerado. Frente a diversos estímulos --que en el caso del SAHS podrían ser hipoxias transitorias, reacciones neurovegetativas o incluso presiones pleurales negativas-- se generan mediadores que pueden alterar la transcripción y provocar una respuesta como la descrita previamente: inflamación, disfunción endotelial, oxidación de lipoproteínas e incluso, frente a células espumosas, enfermedad vascular. La respuesta inflamatoria debe ser equilibrada para no ser nociva o insuficiente. El análisis de los diferentes mediadores descritos estima la presencia o no de inflamación y el tipo predominante.
SAHS e inflamación
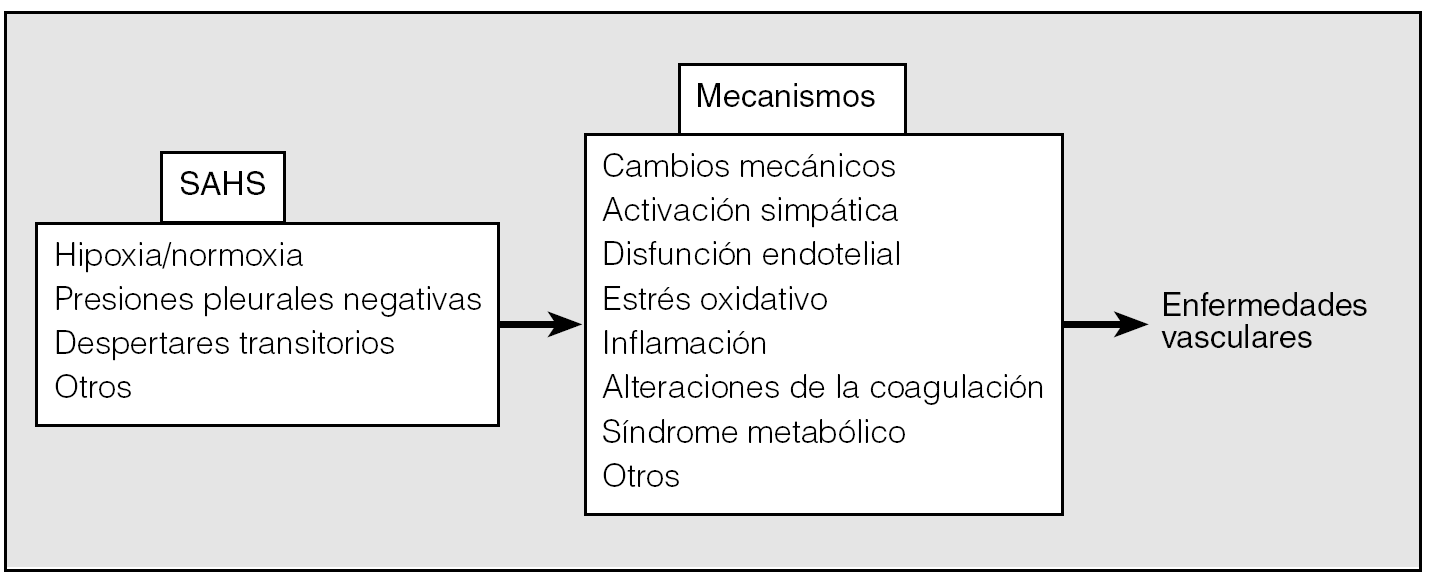
La relación entre el SAHS y la enfermedad cardiovascular puede producirse: a) por los cambios de las presiones intratorácicas, y b) por los cambios inflamatorios, tal como se muestra en la figura 2. Los cambios inflamatorios son complejos, con interrelaciones, y a efectos didácticos se han clasificado muy rígidamente.

Fig. 2. Esquema de los mecanismos por los cuales el síndrome de apneas-hipopneas durante el sueño (SAHS) puede dar lugar a enfermedades cardiovasculares. Los acontecimientos nocturnos originan reacciones diversas, entre ellas la inflamatoria, responsables de la posible afectación vascular.
Cambios mecánicos
Obedecen a la interacción entre la obstrucción de la VAS y las presiones intratorácicas muy negativas, que pueden llegar a ser de 100 cmH2O. Se produce un aumento del gradiente transmiocárdico y, en consecuencia, aumenta de un modo considerable la poscarga. Estas presiones negativas dan lugar a un aumento del retorno venoso, que desplaza el septum hacia la izquierda, con el consiguiente deterioro de la función del ventrículo izquierdo. En resumen, la combinación del aumento de la poscarga por las presiones negativas, y también por la hipertensión que se genera, el desplazamiento del septum y, por último, la hipoxemia que se crea da lugar a una disfunción cardíaca que revierte totalmente con presión positiva continua de la vía aérea (CPAP)4,5.
Anomalías neurovegetativas
En sujetos sanos la actividad simpática durante la noche, la presión arterial y la frecuencia cardíaca disminuyen; en la fase REM, aumentan. Los quimio y barorreceptores controlan los cambios gasométricos y de la presión arterial a través, entre otros, de la activación o reducción de la actividad de los sistemas simpático y parasimpático. Los pacientes con SAHS, por la hipoxemia y el resto de acontecimientos que ocurren durante las apneas, muestran durante la noche y durante el día un incremento de la actividad simpática que incrementa la presión arterial. En comparación con personas sanas, presentan taquicardia, reducción de la variabilidad cardíaca y aumento de la variabilidad de la presión arterial. Todos estos cambios representan un factor de riesgo acusado de daño vascular6.
Disfunción endotelial
El endotelio regula el tono vascular mediante una serie de mediadores (endotelina 1 y tromboxano como vasoconstrictores, y prostaciclina y óxido nítrico como vasodilatadores). Frente a diversos estímulos como los que componen los factores de riesgo cardiovascular (diabetes, tabaco, lípidos) o aquellos que acontecen en el SAHS, tales como hipoxia e hipercapnia, entre otros, se produce una respuesta endotelial anómala que da lugar a vasoconstricción, reducción de la capacidad de vasodilatación y una reacción inflamatoria. Se facilita la arteriosclerosis3,7, se incrementan las moléculas de adhesión y todo ello puede revertir con CPAP8. En el SAHS los episodios de hipoxia a través del factor de inducción de hipoxia dan lugar a un aumento del factor de crecimiento endotelial vascular, que es un elemento que incrementa la vascularización y, en consecuencia, protege. El equilibrio entre los factores previos o la intensidad del estímulo determinará si la respuesta endotelial vasoconstrictora y arteriosclerótica va a desarrollarse con la consiguiente aparición de enfermedad vascular.
Estrés oxidativo
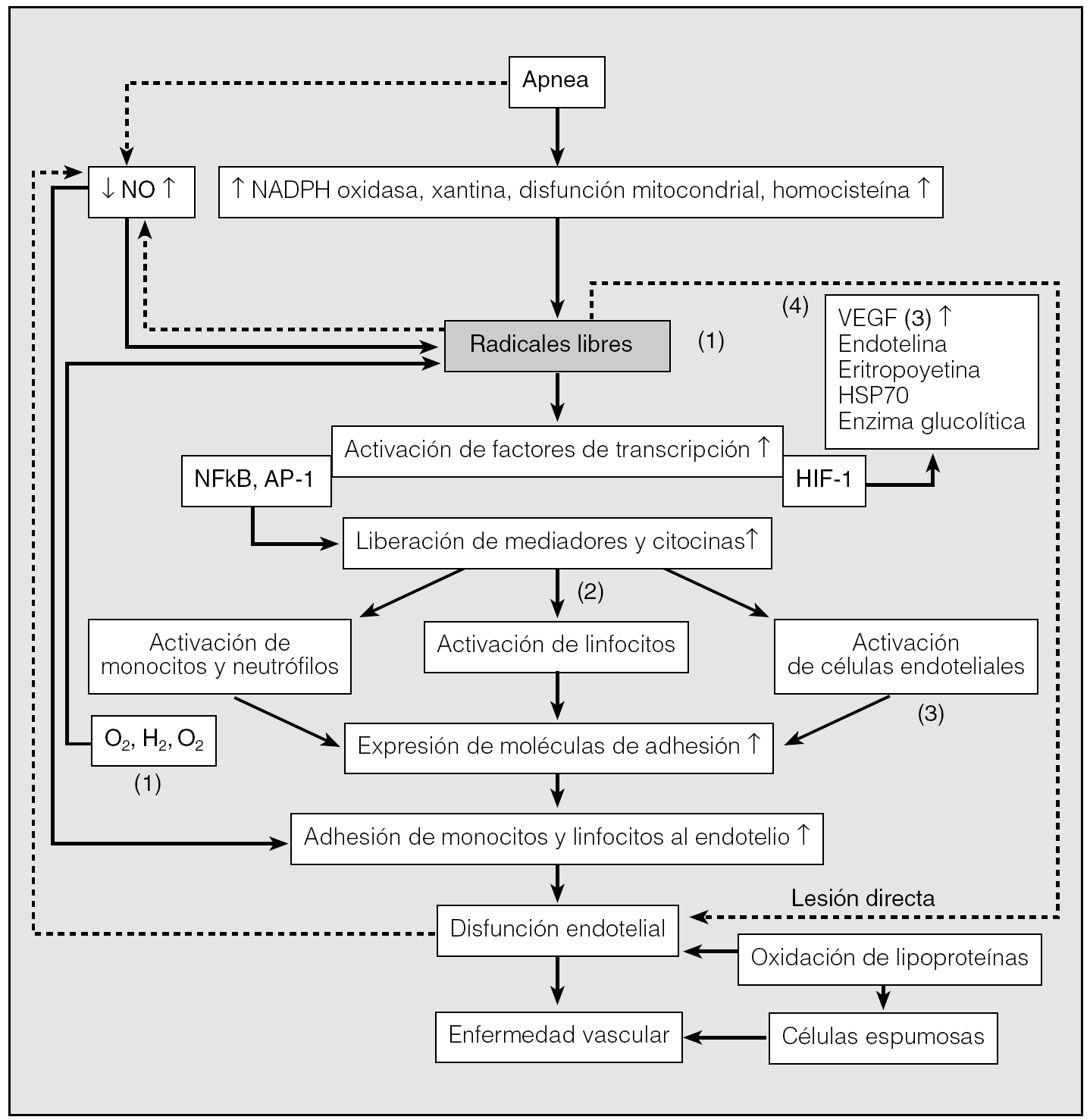
Un radical libre es cualquier molécula con uno o más electrones desapareados La oxidación es el proceso mediante el cual átomos traspasan electrones a otro átomo. Los radicales libres recorren el organismo para obtener un electrón que los estabilice9. Al obtenerlo de una molécula normal convierten a ésta, a su vez, en un radical libre, pero también se considera que la lesionan. Se desencadena una reacción en cadena que daña el organismo. Los radicales libres no son intrínsecamente perjudiciales, pues son necesarios en nuestro cuerpo para luchar contra diversas noxas. Los neutrófilos los producen para atacar los microorganismos y ya en la respiración celular se produce adenosintrifostato y un porcentaje pequeño de radicales libres. Los antioxidantes los regulan. En personas con SAHS se pueden producir cantidades muy notables de radicales libres que los antioxidantes no pueden compensar9. Se produce entonces lesión celular. En el SAHS los radicales libres se originan en las mitocondrias, los fagocitos y el tejido vascular. Los fenómenos que ocurren durante las apneas darían lugar a un aumento de oxidasas, probable disfunción mitocondrial que impulsa un importante incremento de radicales libres y otros mediadores. Éstos dan lugar a una activación de los factores de transcripción, que inducirán anomalías en la expresión génica, con la formación de factores inflamatorios responsables de una cascada inflamatoria global tal como se ha descrito previamente9. Se activan los linfocitos, monocitos, neutrófilos, células endoteliales, las moléculas de adhesión y, además, la angiogénesis, con las consecuencias antes descritas (fig. 3)9.

Fig. 3. Diversos mecanismos que intervienen en la respuesta celular y molecular para que el síndrome de apneas-hipopneas durante el sueño (SAHS) pueda producir enfermedad vascular. Diversos acontecimientos que tienen lugar durante las apneas dan lugar al incremento de radicales libres, se producen anomalías en la transcripción de diversos mediadores, así como una reacción inflamatoria generalizada y daño vascular. NFκB: factor nuclear κB; HIF: factor de inducción de hipoxia; VEFG: factor de crecimiento endotelial vascular; NADPH: nicotinamida adenindinucleótido; AP: proteína activadora. 1: estrés oxidativo; 2: inflamación; 3: disfunción endotelial; 4: angiogénesis.
Inflamación
Se ha demostrado que la inflamación sistémica tiene responsabilidad en la patogenia de la insufiencia cardíaca10. Por la hipoxia y la desestructuración del sueño entre otros, los pacientes con SAHS experimentan una inflamación sistémica con incremento de la proteína C reactiva, factor de necrosis tumoral y citocinas que activan la vía inflamatoria descrita11-13. Se generan moléculas de adhesión que, junto a la cadena inflamatoria, pueden favorecer el desencadenamiento de la arteriosclerosis3. La homocisteína es un aminoácido que se genera a partir de las proteínas de las dieta. Las concentraciones sanguíneas elevadas de homocisteína son de origen genético o aparecen por déficit de ácido fólico o folato. La homocisteína es un factor de riesgo para la arteriosclerosis. Respecto a la enfermedad coronaria, las concentraciones elevadas de homocisteína en sangre inducen un riesgo similar al del tabaquismo o al de la hiperlipemia. Se supone que actúa provocando disfunción endotelial, estrés oxidativo, aumento de la agregación plaquetaria y proliferación de la musculatura lisa. En el SAHS todos los elementos que acontecen durante las apneas por un mecanismo no claro inducen el incremento de homocisteína14.
Síndrome metabólico asociado al SAHS
El síndrome metabólico se define cuando un sujeto tiene al menos 3 criterios de los 5 siguientes: aumento del diámetro de la cintura, hipertensión, hiperglucemia, reducción del colesterol de alta densidad y aumento de los triglicéridos. Se pueden asociar además inflamación, disfunción endotelial, microalbuminuria, resistencia a la insulina y activación simpática. Los pacientes con SAHS poseen la mayoría de los factores citados previamente y un 40% de ellos tienen criterios de síndrome metabólico15,16. Otro elemento relacionado con la obesidad es la leptina, una hormona derivada del tejido adiposo que reduce el apetito y se genera frente a cantidades elevadas de grasa. Produce agregación plaquetaria y actúa sobre otros elementos de riesgo cardiovascular. En el SAHS las concentraciones de leptina están elevadas en comparación con los obesos sin SAHS, lo cual indica resistencia y, en consecuencia, una tendencia a engordar17. Se ha señalado que el SAHS podría inducir síndrome metabólico a través de la hipoxemia, toda una cadena inflamatoria y estrés oxidativo, entre otros, que mejora con CPAP18. Sin embargo, la comorbilidad asociada representa un factor de confusión definitivo hasta el momento16. En la actualidad, al igual que sucedió con hipertensión y SAHS, no está clara la relación de causalidad del SAHS con el desarrollo del síndrome metabólico. Los estudios se hallan en una fase inicial.
Alteraciones de la coagulación
La agregación plaquetaria, las concentraciones de fibrinógeno e incluso la viscosidad sanguínea están elevadas en el SAHS19. Esta circunstancia se atribuye al aumento de la actividad simpática que se origina durante las apneas. Se ha comprobado que los trastornos de la coagulación se reducen con CPAP19. En consecuencia, en el futuro habrá que considerar los efectos nocivos del SAHS y la eficacia de la CPAP, no sólo respecto a la somnolencia y los síntomas clínicos, sino mediante otros parámetros como los que se han descrito. Se precisan trabajos más concluyentes con un elevado número de pacientes para aplicar estos conceptos, que sin duda, en nuestra opinión, van a ser trascendentes para una nueva visión del SAHS.
Inflamación y vía aérea superior
En la última década han aparecido datos que revelan la presencia de marcadores de inflamación en la VAS y que apuntan a que la inflamación podría tener un papel en la fisiopatología del SAHS20-23. En estos pacientes es frecuente el hallazgo macroscópico de inflamación local de la VAS, el aumento del tamaño y el enrojecimiento de la úvula y del paladar blando. Se postula que estos cambios inflamatorios locales son debidos a la vibración generada por el ronquido20. Pero no solamente se han encontrado cambios inflamatorios en las estructuras susceptibles a la vibración del ronquido, sino que los pacientes con SAHS, en ausencia de otros procesos que lo justifiquen, tienen inflamación nasal21, indicios de inflamación en la vía aérea22 y aumento de neutrófilos en el esputo inducido23, así como inflamación en el parénquima pulmonar de un modelo animal de síndrome de apneas en rata24. Todo ello indicaría que las presiones generadas a lo largo de todo el sistema respiratorio durante las obstrucciones de la VAS pueden contribuir a la aparición de inflamación en los pacientes con SAHS. Los estudios histológicos de la VAS en el SAHS se han realizado sobre muestras de uvulopalatofaringoplastia provenientes de pacientes con SAHS, roncadores sin apneas y controles sometidos a otras intervenciones quirúrgicas del área otorrinolaringológica, o bien de muestras de necropsias. Uno de los primeros cambios histológicos descritos en el SAHS fue la presencia de un importante edema subepitelial25. El edema, una de las manifestaciones histopatológicas de inflamación local, está asociado a un aumento de la permeabilidad vascular, por lo que algunos estudios lo han relacionado con congestión y dilatación vascular26. Otras veces, la extravasación de líquido hacia la matriz extracelular puede manifestarse como un aumento del grosor de la lámina propia, indicativo de edema intersticial27. Junto con el edema, se han señalado otras alteraciones histopatológicas en la VAS de pacientes con SAHS, en las que la inflamación parece tener algún papel. Sekosan et al27 describieron la aparición de un extenso infiltrado inflamatorio leucocitario con predominio de células plasmáticas en la lámina propia. Paulsen et al20 detectaron infiltrado inflamatorio en la lámina propia, en este caso con predominio de células T (CD3+), junto con una reducción de la densidad y altura de las estructuras papilares del tejido conectivo que se introducen hacia el epitelio, cuya función es el anclaje estructural y el soporte nutricional. En este estudio los pacientes con SAHS tienen diferencias en el patrón de expresión de algunas citoqueratinas y acantosis en el epitelio. Woodson et al26 también encontraron cambios estructurales como la acantosis en el epitelio, con hipertrofia en las glándulas mucosas, pero al mismo tiempo observaron atrofia focal de fibras musculares y cambios degenerativos en nervios periféricos. En este sentido, Boyd et al28 han descrito un aumento en las terminaciones nerviosas musculares, con evidencia de denervación acompañada de un infiltrado inflamatorio con predominio de células T (CD4+ y CD25+). Por otro lado, Friberg et al29 revelaron la presencia de numerosas terminaciones nerviosas anómalas de aspecto varicoso en la lámina propia, y finalmente Hernández et al30 han correlacionado positivamente la densidad de fibras nerviosas y la infiltración por leucocitos (CD45+) en la mucosa de la úvula de pacientes con SAHS. Todo ello hace pensar en la presencia de una neuropatía, probablemente mediada por inflamación, responsable de los déficit sensoriales detectados en la VAS del SAHS31 y de la disfunción muscular de la VAS, que contribuyen al proceso de obstrucción. Algunas de estas alteraciones también se han identificado en pacientes roncadores sin apneas, aunque en menor grado, pero no en individuos controles no roncadores26,29. Cabría pensar que, en fases iniciales, la vibración del ronquido provocaría modificaciones estructurales e inflamación local en la VAS que iría progresando paulatinamente. La inflamación local per se condicionaría el compromiso anatómico, lo que facilitaría la obstrucción. Aparte del compromiso estructural, la inflamación local desempeñaría un papel en la disfunción neuromuscular de la VAS alterando los reflejos nerviosos y la función de los músculos que controlan el calibre de la vía aérea32. Una vez que los acontecimientos respiratorios obstructivos están presentes, los cambios en las presiones de la vía aérea y la hipoxemia contribuirían a amplificar la inflamación en otras zonas del sistema respiratorio, incluso a nivel sistémico. Se estaría frente a un círculo vicioso en el que a mayor inflamación, mayor disfunción y más obstrucción, que finalmente genera más inflamación. En todo este proceso, la inflamación sería una consecuencia de los fenómenos que acontecen durante los acontecimientos respiratorios. No obstante, es difícil disponer de datos histológicos que demuestren la remisión de estas alteraciones después de un tratamiento efectivo, como la CPAP. Tampoco se puede descartar que se trate de una enfermedad con base inflamatoria que en algún momento se active provocando toda la cascada de alteraciones locales antes descritas. Posiblemente nos encontremos frente a un nuevo campo para definir mejor los mecanismos subyacentes en la fisiopatología del SAHS.
Características mecánicas de los músculos de la vía aérea superior
En la VAS existe un gran número de músculos, con acciones a menudo complementarias y sinérgicas, que pueden modificar el tamaño y la configuración de la faringe. Los principales músculos dilatadores de la faringe son el tensor palatini, el geniohioideo, el esternohioideo y, sobre todo, el geniogloso (GG). Durante la vigilia, el tono muscular basal de los músculos de la VAS mantiene la permeabilidad de la luz y el flujo aéreo. En los enfermos con SAHS, el tono basal diurno del GG está aumentado respecto a los sujetos sanos y es proporcional al grado de resistencia al flujo aéreo intraluminal33. Durante el sueño, al igual que todos los músculos de la VAS, el GG disminuye su grado de actividad basal y también su capacidad de respuesta ante la presión negativa dentro de la luz faríngea generada en cada contracción diafragmática34. De esta forma favorece el colapso de la VAS y la aparición de apneas. Una vez establecida la apnea, el GG incrementa progresivamente su actividad electromiográfica hasta alcanzar un pico, que coincide con la reapertura de la luz faríngea y el restablecimiento del flujo aéreo. Estas observaciones apuntan a que el GG está implicado tanto en la aparición como en la resolución de las apneas. Según Petrof et al35, la hiperactividad muscular mantenida podría inducir cambios adaptativos en los músculos de la VAS, especialmente en el GG. Para poder generar más fuerza, sus fibras musculares deberían hipertrofiarse y, asimismo, aumentar el porcentaje relativo de fibras tipo II, menos resistentes que las de tipo I, pero capaces de generar más fuerza, imprescindible para vencer el colapso de la vía aérea durante las apneas. Estos cambios adaptativos provocarían, no obstante, efectos secundarios indeseables, a saber: la hipertrofia muscular ocupa más espacio, lo cual comprometería la luz faríngea; además, la mayor fuerza generada por las fibras tipo II y su hiperactividad mantenida acabarían por dañar algunas fibras musculares que serían sustituidas por tejido fibroso. Este tejido también ocupa espacio y disminuye la eficacia de la contracción muscular. Todo ello comprometería aún más la luz faríngea, lo que favorecería la tendencia al colapso. En esta línea, Sériès et al36 demostraron que el GG de los enfermos con SAHS tiene una mayor proporción de fibras tipo IIA (respecto a las IIB) que el de los roncadores simples, y que en ellos el músculo de la úvula tiende a fatigarse más y a tener más fibras tipo II que el de los roncadores simples37. Aunque estos resultados tienden a confirmar la teoría de Petrof et al35, hay que tener en cuenta, como limitaciones de estos estudios, que el grupo control estuvo formado por roncadores y que los estudios electrofisiológicos se efectuaron en biopsias de úvula, músculo que no ha demostrado estar especialmente implicado en la fisiopatología del síndrome de apneas obstructivas del sueño.
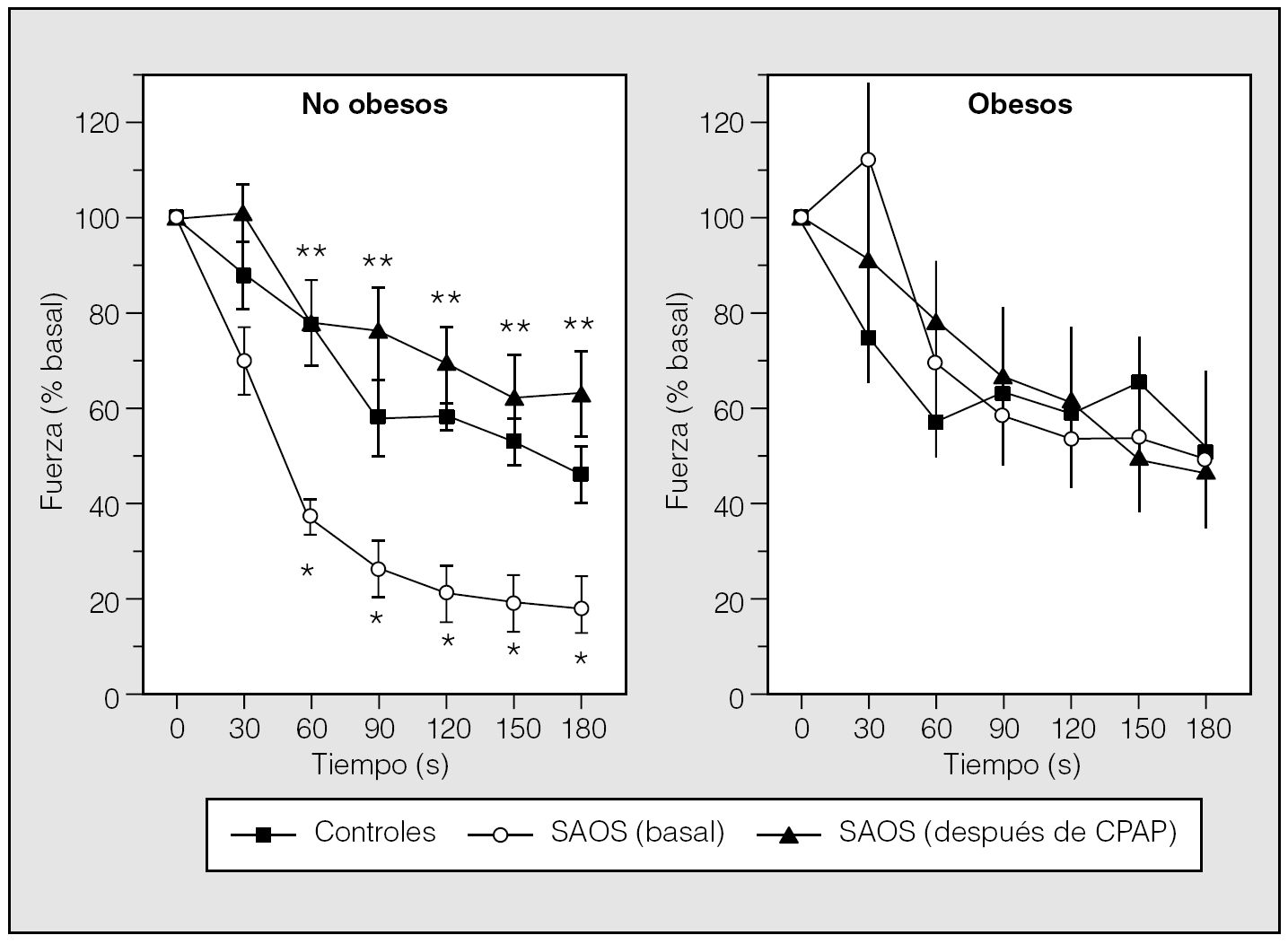
Tratando de evitar estas limitaciones Carrera et al38 han evaluado, in vitro, las propiedades contráctiles e histoquímicas del GG, el principal músculo dilatador de la faringe. Para ello se estudió a un grupo de pacientes con SAHS en el momento del diagnóstico, antes de iniciar ningún tratamiento, un grupo control de sujetos sanos (no roncadores) sin SAHS y un tercer grupo de enfermos con síndrome de apneas obstructivas del sueño tratados con CPAP durante, al menos, un año. Este tercer grupo permitiría estudiar el efecto de la CPAP sobre las propiedades del músculo GG. Las principales observaciones de este trabajo fueron: a) la fatigabilidad in vitro del GG está aumentada en los enfermos con SAHS; b) la causa sería una mayor proporción de fibras tipo II, más potentes pero menos resistentes que las tipo I, y c) la estructura y la función del GG se normalizan después de un año de tratamiento con CPAP. El hecho de que las alteraciones electrofisiológicas e histológicas observadas sean corregibles con CPAP induce a pensar que son una consecuencia de los episodios de colapso de la VAS, no su causa, lo que apoyaría la hipótesis integradora de Petrof et al35 antes comentada. Sin embargo, la obesidad, que es una característica clínica común del SAHS, podría influir tanto en la estructura como en la función de los músculos de la vía aérea. Con el objetivo de evaluar el impacto de la obesidad sobre la estructura y función del GG en pacientes con SAHS, Carrera et al39 ampliaron su estudio inicial mediante la inclusión de más sujetos. Se estudió a los mismos grupos que en el estudio inicial (pacientes en el momento del diagnóstico, controles y otro grupo de pacientes después de un año de tratamiento con CPAP), pero además cada grupo estaba dividido en 2 subgrupos: obesos y no obesos. Los hallazgos de este nuevo estudio fueron: a) se confirmó que, en el momento del diagnóstico, los pacientes con SAHS presentaban un porcentaje más elevado de fibras de tipo II que los controles, independientemente de la obesidad; b) la obesidad no influye en la fuerza máxima de contracción, y c) las características de fatigabilidad del GG eran muy diferentes en los 2 grupos con SAHS estudiados: en los pacientes con SAHS no obesos, el GG mostró una resistencia a la fatiga disminuida, la cual se normalizó totalmente después del tratamiento con CPAP. En los pacientes obesos, no hubo diferencia en fatigabilidad del GG respecto a la observada en controles obesos. Estos resultados indicarían que la obesidad (en ausencia de SAHS) no supone una mayor carga de trabajo sobre el GG ni influye por sí misma en la estructura del GG. La observación más relevante y sorprendente del estudio de Carrera et al39 es que la resistencia del GG está disminuida en los sujetos no obesos con SAHS, al contrario de lo que ocurre en los obesos (fig. 4), y que este efecto en los pacientes no obesos con SAHS es reversible con el tratamiento prolongado con CPAP39. Dado que estos 2 grupos de pacientes comparten la misma distribución anormal de fibra, este hecho no puede explicarse por diferencias estructurales. Por ello, tiene que estar relacionado, necesariamente, con diferencias en los mecanismos celulares involucrados en los procesos de contracción muscular y generación de fuerza, como la liberación y toma de calcio y adenosintrifosfato, así como la síntesis y liberación de fosfocreatinina, entre otros. El hecho de que la resistencia del GG sea anormal y corregible con CPAP sólo en pacientes con SAHS no obesos puede tener implicaciones clínicas. Sobre la base de estas diferencias mecánicas entre pacientes obesos y no obesos se pueden plantear, al menos teóricamente, diferentes estrategias terapéuticas. Por ejemplo, el conocimiento de los mecanismos celulares que regulan la capacidad de resistencia puede llevar a tratamientos farmacológicos mejor orientados a este objetivo. Por otra parte, el hecho de que la CPAP normalice la resistencia del GG en pacientes no obesos podría permitir, en teoría, plantear la posibilidad de tratar a los pacientes con síndrome de apneas del sueño mediante CPAP de forma intermitente. Este planteamiento puede tener un impacto clínico importante, ya que en algunos laboratorios el 50% de los pacientes con SAHS no son obesos. En resumen, los músculos de la VAS de los pacientes con SAS presentan una alteración en la distribución del tipo de fibras musculares, con un predominio de las fibras tipo II, más potentes pero menos resistentes que las tipo I. La obesidad influye en la mecánica de la vía aérea de forma que las alteraciones contráctiles del GG sólo se aprecian en pacientes no obesos.

Fig. 4. Evolución a lo largo del tiempo de la fuerza del geniogloso (media ± desviación estándar) como respuesta a una estimulación eléctrica repetida (40 Hz) en sujetos no obesos y obesos. CPAP: presión positiva continua de la vía aérea; SAOS: síndrome de apneas obstructivas del sueño; *p < 0,05 entre obsesos sin tratamiento y controles; **p < 0,05 entre pacientes antes y después de CPAP. (Modificada de Carrera et al39.)
El SAHS es una enfermedad muy común que, aparte de los síntomas clásicos como la somnolencia, tiene otras implicaciones notorias en, por ejemplo, el síndrome metabólico o las enfermedades cardiovasculares, mediada, entre otros, por la inflamación. Es de esperar que el conocimiento de los mecanismos implicados en la VAS del SAHS permita tratamientos etiológicos más definitivos y menos engorrosos. Es posible que, en pocos años, una muestra de sangre para medir una serie de biomarcadores sea muy útil a efectos de valoración, seguimiento e incluso diagnóstico.







