

Alpha-1 antitrypsin deficiency (AATD) is a genetic disease with a prevalence of about 1:5000 individuals in Italy.1–4 This deficiency predisposes to pulmonary emphysema while polymerogenic mutations can lead to retention of AAT in the liver, causing liver diseases.5 A diverse range of mutations within the SERPINA1 gene contributes to a growing array of AAT variants. The alleles were named with the prefix protease inhibitor* (Pi*) serving as an alias for the gene.1 The most common (normal) allele is Pi*M, and the most common pathogenic alleles are Pi*Z and Pi*S and common AAT genotypes are MM, MS, MZ, SS, SZ, ZZ.6 Apon suspicion of AATD, it is necessary the measurement of plasma AAT levels via nephelometry accompanied by an assessment of C-reactive protein, since AAT is an acute-phase reactant that increases during infection or inflammation.7 However, plasma level alone is an insufficient parameter for the diagnosis, therefore, phenotyping, genotyping or even gene sequencing are needed in inconclusive cases.8,9 This study aims to conduct a comprehensive analysis of the epidemiology of AATD in the central-south region of Italy, using samples referred by pulmonologists and hepatologists to a designated genetic laboratory. The primary objective is to describe the identified genotypes and categorize them based on associated diseases.
1706 participants with a clinical suspicion of AATD (i.e., respiratory symptoms or family history) recruited through a screening program (supplementary material), in different Italian regions of Centre-Southern Italy (Campania, Calabria, Lazio, Sicily, and Puglia), between 2017 and 2021, were included in the study (Fig. 1).

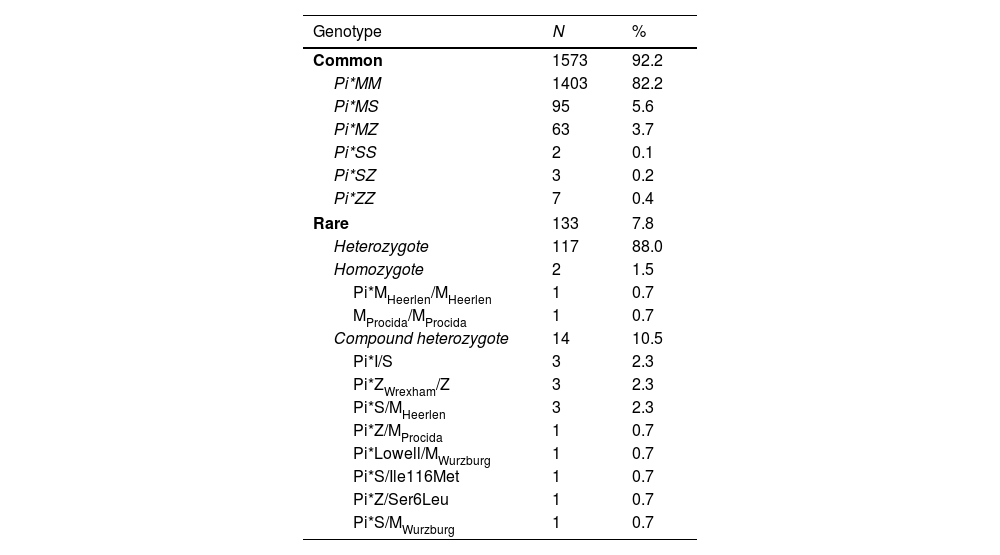
Of the sample, 61.2% were male and 38.8% female. 1490 subjects (87.3%) had pulmonary phenotype, 73 (4.3%) had hepatic and pulmonary disorders, 124 (7.3%) had a first degree relative with AATD and 19 (1.1%) had “mixed lung disease” (see Table S2). Overall, 1403 (82.2%) subjects presented a Pi*MM genotype, 95 (5.6%) Pi*MS, 63 (3.7%) Pi*MZ, 2 (0.1%) Pi*SS, while 3 (0.1%) Pi*SZ, and 7 (0.4%) Pi*ZZ; 7.8% (n=133) of the population had rare pathogenic variants. Among these, the majority were heterozygous (n=117; 88%), 14 were compound heterozygous (10.5%) and only 2 homozygous genotypes were identified (1.5%): Pi*MHeerlen/MHeerlen (found in Puglia) and Pi*MProcida (found in Sicily) (Table 1). Analysis of clinical manifestations and genotypes revealed COPD with/without pulmonary emphysema as the most prevalent conditions (934/1706), followed by bronchiectasis and asthma (Table S2). Furthermore, the most common liver manifestations were hepatitis, liver cirrhosis, and chronic liver disease, affecting 73 patients. In addition, we collected the diseases for which genetic analysis was requested and rated their distribution by region, as shown in Fig. S2. By dividing population: patients<18 years were classified as Group A (n=27), 18–65 years as Group B (n=996) and >65 years as Group C (n=671). Instead, 25% of Group A (7/27) had AATD. MM genotype was found in more than 80% of Group B and C, followed by a rare variant (5%) and Pi*MS. Conversely, about 50% of Group A had the Pi*MM genotype, followed by the Pi*MZ genotype (18.5%), a rare variant (11%) and Pi*MS (7%). SERPINA1 variants were more frequent in males with a statistical difference for Pi*MM (p=0.008), Pi*MS (p=0.03) and the Pi*MMalton (p=0.03), while the other rare variants were equally distributed in males and females (p=NS). Approximately 28% of patients (n=478) were subjected to a quantitative measurement of AAT serum value. Pi*MM and Pi*MS were associated with normal mean serum values of AAT (n.v. 100–220mg/dL), 137.2mg/dL and 125.3mg/dL, respectively, and as expected, Pi*SZ and Pi*ZZ genotypes displayed the lowest values, 47.5mg/dL and 36.8mg/dL, respectively.
Alpha-1 antitrypsin deficiency (AATD) genotypes of the study population.
| Genotype | N | % |
|---|---|---|
| Common | 1573 | 92.2 |
| Pi*MM | 1403 | 82.2 |
| Pi*MS | 95 | 5.6 |
| Pi*MZ | 63 | 3.7 |
| Pi*SS | 2 | 0.1 |
| Pi*SZ | 3 | 0.2 |
| Pi*ZZ | 7 | 0.4 |
| Rare | 133 | 7.8 |
| Heterozygote | 117 | 88.0 |
| Homozygote | 2 | 1.5 |
| Pi*MHeerlen/MHeerlen | 1 | 0.7 |
| MProcida/MProcida | 1 | 0.7 |
| Compound heterozygote | 14 | 10.5 |
| Pi*I/S | 3 | 2.3 |
| Pi*ZWrexham/Z | 3 | 2.3 |
| Pi*S/MHeerlen | 3 | 2.3 |
| Pi*Z/MProcida | 1 | 0.7 |
| Pi*Lowell/MWurzburg | 1 | 0.7 |
| Pi*S/Ile116Met | 1 | 0.7 |
| Pi*Z/Ser6Leu | 1 | 0.7 |
| Pi*S/MWurzburg | 1 | 0.7 |
The prevalence of different genotypes identified in the study population was categorized into common genotype 1573/1706 (92.2% overall) and rare 133/1706 (7.8% overall). Subsequently, the rare genotypes were further classified based on whether they included a rare variant, with distinctions made for those possessing only one copy of a rare allele (heterozygous), two copies (homozygous), and two distinct copies of a rare allele (compound heterozygote).
Scientific evidence has demonstrated that AATD is the main known genetic risk factor for COPD, and it is estimated that up to 3% of patients diagnosed with COPD may have AATD.10,11 In this line, our study showed that 62.6% patients with pulmonary phenotype were affected by COPD. Not to be underestimated, is the close relationship between AATD and bronchiectasis with a prevalence of 3.8–73.1%12 and the association between AATD and asthma with a rising prevalence of 4–38%.16 This may be due the immune-modulatory effects of AAT at the bronchial level.13,14 In our study, Pi*MM was the most represented genotype (82%), followed by MZ and MS genotype. Nevertheless, Pi*MZ genotype is especially important when it comes to current or ex-smokers, as the decline of lung function and an increased risk for emphysema development is high in these individuals.15 Their detection has an utmost importance in smoking prevention/cessation, minimizing the hazards of occupational respiratory pollutants, the opportunities to receive augmentation therapy, and the potential for family planning and guided genetic counseling/testing.16 In addition, 124 patients presented a family history for emphysema and/or first-degree relatives with AATD, and 62.1% of them had at least one heterozygous AATD confirming the heritability of the disease. Not only, 25% young subjects had AATD. This underscores the necessity for individuals with a family history of respiratory illness or early symptoms that young adults might overlook to undergo testing for AATD, emphasizing the significance of heterozygotes. In contrast, Pi*ZZ genotype was identified in only 7 (0.4%) participants, and Pi*SZ genotype only in 3 (0.1%). It is well known that the prevalence of the PI*Z gene in Europe is higher in northwestern countries, with a decreasing gradient toward the south-east.17 This gradient was respected in Italy, where most Pi*ZZ individuals in our series were found in northern areas. Interestingly, 7.7% (n=131) of the participants had rare pathogenic variants, a significant higher prevalence compared to Pi*ZZ genotype. The most frequent appears to be Pi*MMWürzburg (17/131), followed by Pi*MPLowel (13/131) and then Pi*MMProcida (10/131). M-like variants appear to be mostly affected at respiratory level, with a strong association to cigarette smoke exposure. In fact, early and accurate detection of “rare” variants gives the affected individual the opportunity to make lifestyle changes.18 The diagnosis of AATD typically occurs around the age of 43–45 years and progresses more rapidly in heavy smokers or individuals exposed to significant air pollution.19,20 However, it often remains under-diagnosed, with significant delays from the onset of initial symptoms to diagnosis, estimated at 5–8 years.4 This aligns with the average age of enrollment of participants in our study (56 years). Hence, to mitigate this delay, international societies recommend testing for AATD in individuals diagnosed with COPD, adult asthma with incomplete reversibility, bronchiectasis, unexplained liver disease, panniculitis, or C-ANCA vasculitis.19 Furthermore, the epidemiology of AATD remains largely undisclosed in numerous countries, primarily due to under-diagnosis and the absence of registries for the identified patients. The international EARCO registry, however, is actively tackling this challenge by enlisting centers worldwide and advocating for research in AATD.20
Our study presents some limitations: lack of AAT quantitative assays compared to the number of genetic analyses conducted and a lack of clinical information; the lower it yields epidemiological data but on a very selected group of patients.
In conclusion, the importance of testing for AATD extends beyond identifying homozygotes such as Pi*ZZ with the most severe clinical manifestations but it is crucial for identifying heterozygotes and, notably, individuals with rare variants who exhibit near-normal levels of AAT in their blood but face a heightened risk of developing lung disease.
FundingThis research received no external funding.
Authors’ contributionsConceptualization, G.S., R.S., M.P.F.B., M.M. and D.L.; methodology, G.S., R.S., M.P.F.B., A.L., M.M. and D.L.; validation, G.S., R.S., M.P.F.B., M.M. and D.L.; formal analysis, P.T.; resources, G.S., R.S., P.T., P.S., E.G., M.F.D.A, A.L., L.P., A.H., G.M., M.P.F.B., M.M. and D.L.; data curation, G.S., R.S., P.T., M.P.F.B., M.M. and D.L.; writing-original draft preparation, G.S., R.S., A.H and P.T.; writing-review and editing, G.S., R.S., M.P.F.B., M.M. and D.L.; visualization, P.T.; supervision, M.P.F.B., M.M. and D.L.; project administration, M.M. and D.L. All authors have read and agreed to the published version of the manuscript.
Institutional review board statementThe study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Polyclinic “Riuniti” University-Hospital of Foggia (protocol code 17/CE/2014).
Informed consentInformed consent was obtained from all subjects involved in the study.
Conflicts of interestThe authors declare no conflict of interest.
The followings are the supplementary data to this article:







