

La fibrosis quística es una enfermedad autosómica recesiva monogénica, de la que se han descrito ya más de 1.900 mutaciones agrupadas en 6 clases y que constituye un ejemplo de lo que podría ser una enfermedad bien situada para poder beneficiarse de la medicina personalizada. En la actualidad, 2 enfoques muy diferentes tienen por objetivo corregir el defecto básico: la terapia génica, dirigida a corregir la alteración genética, y la terapia encaminada a corregir el defecto a nivel de la proteína CFTR. Esta última comienza a dar resultados prometedores con diversas moléculas en desarrollo. Ataluren (PTC124) es una molécula diseñada para que los ribosomas se vuelvan menos sensibles a los codones de parada prematuros responsables de las mutaciones clase i. Lumacaftor (VX-809) es un fármaco corrector que está dirigido a mutaciones de clase ii, entre las que figura la más frecuente (Phe508del), con prometedores resultados. Ivacaftor (VX-770) es un fármaco potenciador, el único comercializado hasta el momento, que ha demostrado una buena eficacia para la mutación de clase iii, Gly551Asp, en niños mayores de 6 años y adultos. Además, diversos ensayos están probando estos fármacos o la combinación de ellos para otras mutaciones genéticas menos frecuentes. En los últimos 5 años, la CFTR ha sido designado como una diana terapéutica. Ivacaftor es el primer fármaco que trata el defecto básico de la fibrosis quística, pero solo da respuesta a un escaso porcentaje de los pacientes. Se precisan nuevos fármacos capaces de restaurar la proteína CFTR causada por mutaciones más comunes.
Cystic fibrosis is a single gene, autosomal recessive disorder, in which more than 1,900 mutations grouped into 6 classes have been described. It is an example a disease that could be well placed to benefit from personalised medicine. There are currently 2 very different approaches that aim to correct the basic defect: gene therapy, aimed at correcting the genetic alteration, and therapy aimed at correcting the defect in the CFTR protein. The latter is beginning to show promising results, with several molecules under development. Ataluren (PTC124) is a molecule designed to make the ribosomes become less sensitive to the premature stop codons responsible for class i mutations. Lumacaftor (VX-809) is a CFTR corrector directed at class ii mutations, among which Phe508del is the most frequent, with encouraging results. Ivacaftor (VX-770) is a potentiator, the only one marketed to date, which has shown good efficacy for the class iii mutation Gly551Asp in children over the age of 6 and adults. These drugs, or a combination of them, are currently undergoing various clinical trials for other less common genetic mutations. In the last 5 years, CFTR has been designated as a therapeutic target. Ivacaftor is the first drug to treat the basic defect in cystic fibrosis, but only provides a response in a small number of patients. New drugs capable of restoring the CFTR protein damaged by the most common mutations are required.
La finalización del proyecto del genoma humano supuso una meta relevante para el conocimiento médico, permitiéndonos tener la información necesaria para poder conocer las características únicas de cada individuo1. La consecuencia lógica de este conocimiento sería poder aplicar pruebas diagnósticas y tratamientos concretos a cada paciente basándose en su información genética individual. Esta nueva forma de hacer medicina constituye la denominada medicina personalizada2. Sin embargo, a pesar del gran avance que ha supuesto el conocimiento del genoma humano, su traducción en diagnósticos o tratamientos personalizados ha sido menor de lo esperado. Por el momento, 2 grandes iniciativas, la biología de sistemas3 y la farmacogenética4, están dando pasos en este sentido. El objetivo final de estas iniciativas es conseguir hacer una medicina que sea personalizada a las características de cada individuo, que sea capaz de predecir la aparición o el curso de una enfermedad concreta, que, por tanto, permita establecer estrategias adecuadas de prevención y que, finalmente, permita al paciente ser partícipe de las decisiones. Esto es lo que se ha denominado medicina P45.
La fibrosis quística (FQ) continúa siendo la enfermedad genética más frecuente y letal de la raza blanca. Tiene una incidencia de 1 por cada 2.500-6.000 recién nacidos, dependiendo de la región y de la etnia de origen, y un porcentaje de portadores sanos también variable entre 1:20-376. En España, gracias a la progresiva implantación de los programas de cribado neonatal en las distintas comunidades, se está reconociendo una incidencia inferior a la estimada con anterioridad, siendo en 2009 de: 1/4.430 recién nacidos vivos en Galicia, 1/4.339 en Castilla y León, 1/5.376 en Murcia, 1/5.840 en Cataluña y 1/6.602 en Baleares7. Se estima que en todo el mundo hay unas 70.000 personas que padecen FQ8. La enfermedad se produce por mutaciones en el gen que codifica la denominada proteína reguladora de la conductabilidad transmembrana de la FQ (CFTR, por sus siglas en inglés: cystic fibrosis transmembrane conductance regulator) que actúa como canal de cloro y participa en la liberación de adenosina trifosfato y en la regulación de otros canales de transporte de iones. Esta proteína se expresa en las células epiteliales del aparato respiratorio, en el páncreas, en las vías biliares, en las glándulas sudoríparas y en el sistema genitourinario. Su alteración conduce a una anomalía en el transporte de iones, de manera que los pacientes producen un moco espeso y viscoso, que obstruye los conductos del órgano donde se localiza y con repercusiones multisistémicas que condicionan la variedad de sus manifestaciones clínicas. A pesar de los grandes avances en el tratamiento de la FQ que han conseguido prolongar la supervivencia (mediana estimada actual de 37,5 años)9, aún existe mucho camino por recorrer hasta conseguir que los pacientes con FQ tengan una vida en cantidad y calidad superponibles a sujetos sin la enfermedad. En este contexto, son necesarios nuevos tratamientos que disminuyan la morbilidad y aumenten la supervivencia.
La FQ constituye un ejemplo de lo que podría ser una enfermedad bien situada para poder beneficiarse de la medicina personalizada. Por un lado, se trata de una enfermedad monogénica, en la que una alteración sobre un gen bien identificado es la responsable de su aparición. Además, tiene una fisiopatología bien conocida con dianas terapéuticas bien identificadas. Finalmente, el diagnóstico de la enfermedad precisa de test genéticos que identifican la variedad de la enfermedad, permitiendo identificar el tipo de defecto genético en cada caso concreto10.
En la actualidad, 2 enfoques muy diferentes tienen por objetivo corregir el defecto básico: la terapia génica, dirigida a corregir la alteración genética, y la terapia con moléculas cuyo objetivo es corregir el defecto funcional a nivel de la proteína. La terapia génica está explorando la forma de introducir copias normales del gen en las vías respiratorias de los pacientes con FQ. Consiste en la inserción de un vector recombinante viral al que se le extrae su ADN y se sustituye por el nuevo ADN terapéutico, de manera que este vector viral sirva de vehículo para insertar el ADN en la célula diana. Hasta el momento se han usado diversos tipos de virus como adenovirus o lentivirus. Además, también se han desarrollado partículas no virales como nanopartículas con ADN con capacidad para insertar ADN11. Sin embargo, los resultados por el momento han sido pobres debido a que la duración de la expresión del gen introducido era corta con ambos tipos de vectores12. El UK Gene Therapy Consortium está desarrollando un ensayo clínico en fase 2 para evaluar la eficacia clínica de un vector optimizado de ADN plasmídico/liposoma13. El objetivo de reclutamiento son 130 pacientes y los resultados se esperan para 2014 (NCT01621867)14.
Por otro lado, la terapia dirigida a la restauración de la función de la proteína CFTR ha tenido más éxito. En los últimos años se ha comenzado a tener resultados sobre fármacos capaces de actuar directamente sobre la proteína CFTR. De hecho en enero de 2012 se comercializó en EE. UU. Unidos el primer fármaco capaz de corregir el defecto de las mutaciones Gly551Asp. En las próximas líneas revisaremos la información disponible sobre los avances en medicina personalizada para la FQ y los tratamientos disponibles encaminados a corregir el defecto causante de la enfermedad a nivel de proteína. En la presente revisión, la nomenclatura utilizada para la descripción de las mutaciones del gen CFTR es la desarrollada por la Human Genome Variation Society15.
Mutaciones y defecto proteicoLa FQ es una enfermedad hereditaria autosómica recesiva, por lo que es necesario presentar la mutación en ambas copias del gen CFTR para estar afectado por la misma. Hasta el momento se han identificado más de 1.900 mutaciones del gen CFTR asociadas a la enfermedad, tanto en la secuencia codificadora, como en el ARN mensajero o en otras regiones. Las mutaciones del gen CFTR se encuentran disponibles para consulta en la Cystic Fibrosis Mutation Database16. La primera mutación descrita, y la más frecuente a nivel mundial, es la Phe508del, pero existen otras mutaciones específicas cuya frecuencia varía entre los distintos grupos étnicos. En España se ha descrito una frecuencia media de la Phe508del de entre 50-60% del total de los cromosomas estudiados, siendo la segunda en frecuencia la Gly542X con un 4-8%, seguida por la Asn1303Lys con un 2-4%. Las mutaciones descritas en la actualidad se clasifican en 6 tipos o clases en función del mecanismo por el que causan la enfermedad17. Estos tipos de mutaciones están resumidos en la figura 1. Las mutaciones de clase i conducen a un codón de parada prematuro en el ARN mensajero que impiden que la proteína se traduzca al completo. Por tanto, la proteína producida es corta y no funcionante. Las mutaciones de la clase ii codifican una proteína mal plegada estructuralmente anormal y que se elimina por el retículo endoplásmico antes de llegar a la superficie de la célula. A este grupo pertenece la mutación más común en la FQ, la Phe508del. En las mutaciones de las clases iii a vi las proteínas llegan a la superficie de la célula, pero no funcionan adecuadamente. En el caso de las mutaciones de clase iii tienen disminuida la activación del canal y permanecen cerradas. Las mutaciones de clase iv provocan una disminución de la conductabilidad de iones a través del canal. Las mutaciones clase v codifican proteínas en menor cuantía que resulta en una cantidad reducida de CFTR en la superficie celular, por lo que se produce una cierta función, pero a un nivel disminuido. Finalmente, las mutaciones de la clase vi conducen a una vida media acortada debido a la inestabilidad de la proteína y también pueden dañar la regulación de los canales vecinos a la CFTR en la superficie celular.
Tratamientos modificadores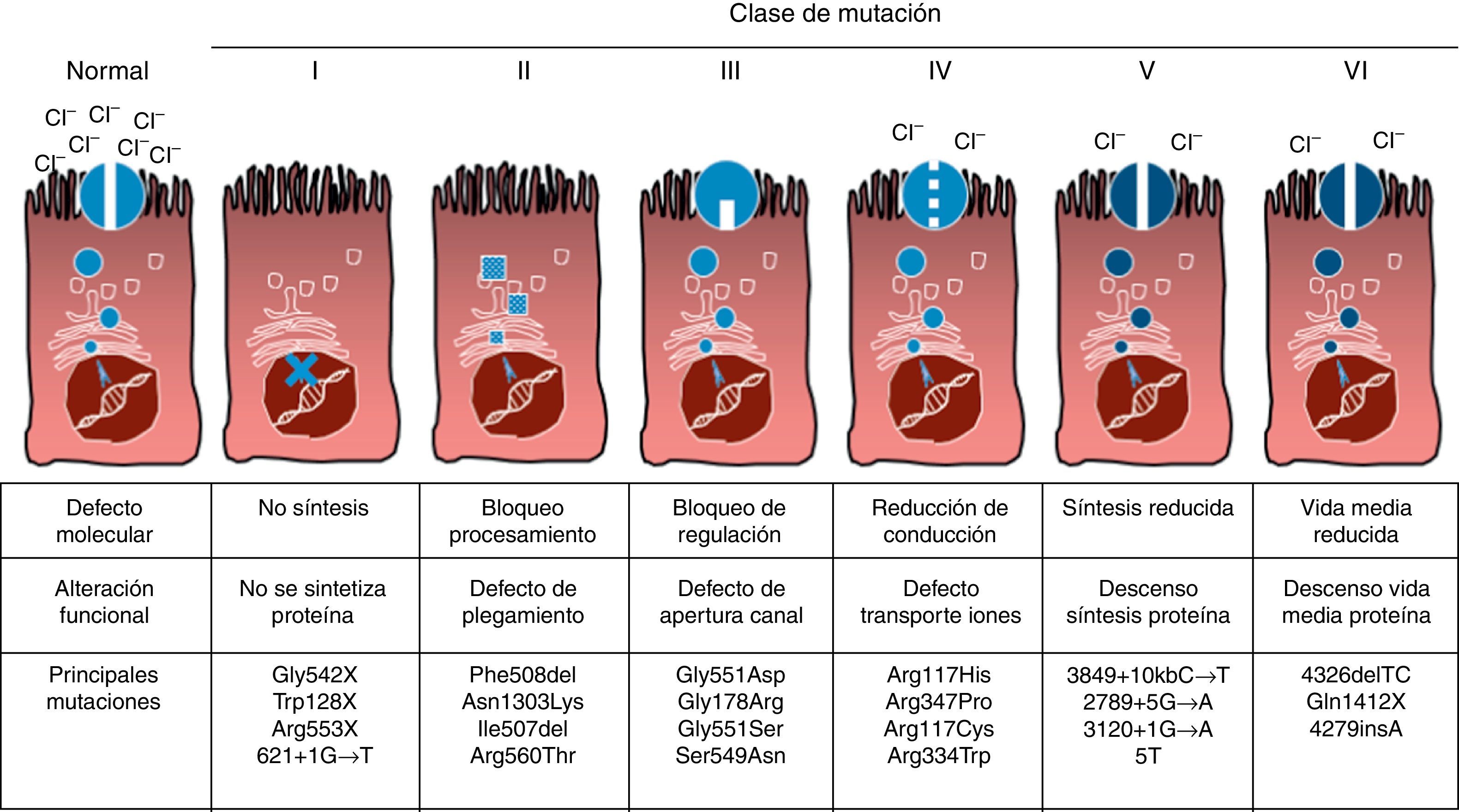
Dentro del desarrollo de los fármacos reparadores de la proteína CFTR, se han identificado 3 grupos principales11. En primer lugar, los supresores del codón de parada prematuro (mutación de clase i). Estos fármacos consiguen que no se identifique este codón de parada prematuro, por lo que la proteína puede seguir su síntesis al completo. En segundo lugar, los denominados fármacos correctores del CFTR. Estos fármacos están diseñados para corregir el tráfico de la proteína con defectos en el plegamiento (mutación de clase ii) hasta la membrana celular donde podría hacer su función casi con normalidad. En tercer lugar, los denominados potenciadores del CFTR. Estos fármacos tienen por diana la proteína CFTR que está en la superficie celular, con objeto de mejorar su función. Los fármacos potenciadores pueden actuar por tanto sobre las mutaciones de clases iii, iv, v y vi. Actualmente, están en investigación numerosas moléculas con uno u otro mecanismo, habiendo llegado ya a comercializarse una de ellas. Ivacaftor (VX-770) es un potenciador del CFTR aprobado en EE. UU. en enero de 2012 para el tratamiento de la FQ en pacientes mayores de 6 años de edad que tienen una mutación Gly551Asp.
Tratamientos para las mutaciones de clase iAproximadamente un 10% de los casos de FQ son debidos a mutaciones de clase i. Para esta clase de mutaciones fueron los aminoglucósidos los primeros fármacos utilizados. Hace varios años, se describió que la gentamicina posee la capacidad de enmascarar el codón de parada prematuro que evita la síntesis de la proteína CFTR. Esto lo consigue mediante la inserción de un aminoácido que permite a los ribosomas continuar la lectura del gen produciendo una proteína de longitud completa. Estudios preclínicos demostraron que se podía sintetizar la proteína en un modelo animal de ratón y que in vitro se recuperaba un 35% de la función de la proteína18,19. El efecto de la administración intravenosa de gentamicina en pacientes con FQ fue evaluado en 2 estudios con pacientes con diversos tipos de mutaciones clase i. Uno con 5 pacientes realizado en EE. UU.20 y otro en 18 pacientes realizado en Francia21. Sin embargo, aunque la respuesta fue positiva, se observó una considerable variabilidad en los resultados, de manera que el beneficio no era universal. Además, la toxicidad de los aminoglucósidos aportaba un perfil desfavorable.
Una alternativa sintética la constituye el ataluren (PTC124; PTC Therapeutics, South Plainfield, NJ, EE. UU.). Ataluren es una molécula diseñada para que los ribosomas puedan leer la información genética «saltándose» el codón de parada prematuro y dando como resultado una proteína CFTR funcional22. La actividad farmacocinética de ataluren ha sido demostrada en modelos animales y en estudios clínicos fase ii. En 2 pequeños estudios iniciales23,24, un grupo de pacientes con FQ tratados con ataluren vía oral mostraron una mejoría de las alteraciones electrofisiológicas de la enfermedad, incrementando el número de células nasales que expresaban la proteína en su superficie. Posteriormente, otro pequeño estudio con 19 pacientes evaluó diferentes dosis de ataluren administrado vía oral cada 8h, observando mejorías en la actividad CFTR y en parámetros clínicos con un buen perfil de seguridad25. En la Conferencia Americana de FQ de 2012 se comunicaron los resultados del ensayo clínico en fase III de ataluren, que aún no han sido formalmente publicados. Un total de 238 pacientes mayores de 6 años fueron aleatorizados para tomar ataluren 10, 10, 20mg/kg o placebo cada 8h durante 48 semanas. No hubo diferencias significativas en el FEV1 de ataluren frente a placebo a las 48 semanas de tratamiento (-2,5% ataluren vs. -5,5% placebo, p=NS). Cuando se hizo una estratificación por el uso de antibiótico crónico nebulizado, la diferencia en aquellos pacientes que no realizaban tratamiento con tobramicina fue de un 6,7% de cambio medio a las 48 semanas a favor de ataluren, mientras que no existía ninguna diferencia en el cambio del FEV1 en aquellos que recibían tobramicina nebulizada. Algo similar ocurrió con el porcentaje de exacerbaciones, en las que no se obtuvo una diferencia significativa entre ambos grupos. Cuando se estratificaron, en los pacientes que no tomaban tobramicina nebulizada el porcentaje de descenso de exacerbaciones fue de un 43% en el grupo de ataluren frente a placebo. Las diferencias del potencial nasal y del cloro en sudor no mostraron diferencias entre ambos grupos recibieran o no antibioterapia nebulizada. Los autores concluyeron que los beneficios son mayores en aquellos enfermos que no recibían antibioterapia crónica nebulizada con aminoglucósidos, especulando que la tobramicina y el ataluren interactúan ambos a nivel ribosómico por lo que pudieran tener una acción antagónica sobre el mismo si se administran a la vez26.
Tratamientos para las mutaciones de clase iiLas mutaciones de clase ii están presentes en un elevado número de los pacientes con FQ, puesto que incluyen la mutación más frecuente de la enfermedad (Phe508del). Este hecho las convierte en un objetivo primordial de la investigación en FQ. Se han estudiado diversas moléculas, la mayoría desarrolladas por Vertex Pharmaceuticals Inc. El primer compuesto corrector, lumacaftor (VX-809), mostró una buena eficacia in vitro, mejorando un 14% el transporte de cloro27. Sin embargo, los resultados fueron algo desalentadores en pacientes, debido a que las mejorías en la concentración de cloro en sudor era muy pequeña (7mmol/L) y sin cambios en el potencial nasal28.
Por otro lado, se han investigado los efectos de la molécula potenciadora del CFTR ivacaftor (VX-770), un fármaco para las mutaciones de clase iii (ver descripción más abajo), en pacientes homocigotos para Phe508del. Los resultados del estudio DISCOVER muestran que ivacaftor no se asocia con una mejoría significativa del FEV1, de la calidad de vida ni del número de exacerbaciones frente a placebo29.
Dado que lumacaftor puede ayudar al movimiento de Phe508del CFTR a la superficie de la célula e ivacaftor aumenta el tiempo de apertura y la conducción de cloro a través de la célula epitelial, la mejoría del defecto Phe508del subyacente podría ser posible con la combinación de ambas moléculas. Los estudios in vitro de lumacaftor e ivacaftor asociados en epitelios respiratorios con mutación Phe508del han mostrado que lumacaftor aislado aumenta el transporte de cloro del CFTR un 15%, y además si se le añade ivacaftor el transporte aumenta a casi el 30%. Esta combinación de medicamentos se ha investigado en un estudio de fase II en pacientes con la mutación Phe508del. Los resultados completos no han sido publicados, pero los datos iniciales sugieren un efecto beneficioso sobre la función pulmonar en Phe508del para homocigotos, aunque no para heterocigotos30. Actualmente, se están desarrollando 2 ensayos clínicos en pacientes de 12 años o mayores homocigóticos para la mutación Phe508del para valorar el tratamiento de lumacaftor junto con ivacaftor; son los estudios TRAFFIC (NCT01807923)31 y TRANSPORT (NCT01807949)32. Los resultados podrían permitir el tratamiento de aproximadamente la mitad de la población con FQ con terapia moduladora del CFTR. Por otro lado, se necesitan estudios para evaluar el efecto de la terapia combinada en individuos Phe508del heterocigotos.
Otro compuesto corrector alternativo es el VX-661 cuyos estudios están actualmente en desarrollo. Su eficacia se está ensayando de manera aislada y combinado con ivacaftor (VX-770) y los resultados estarán pronto disponibles (NCT01531673)33.
Tratamientos para las mutaciones de clase iiiIvacaftor (VX-770) es un potenciador de la proteína CFTR que modula la función de la proteína anormal34. Esta molécula fue originalmente diseñada como un potenciador de la función de CFTR en cultivo de epitelio respiratorio de células que llevan una sola mutación Gly551Asp34. Es el primer fármaco que ha sido aprobado en EE. UU. y en Europa para el tratamiento de la FQ en pacientes con la mutación Gly551Asp. Tras demostrar in vitro la capacidad de ivacaftor para mejorar el transporte de cloro a través de la membrana celular, se realizaron los primeros ensayos en fase ii con 39 pacientes35. En este estudio, la función de la proteína CFTR mejoró a los 3 días de comenzar el tratamiento, llegando la concentración de cloro en sudor hasta el límite de la normalidad. A partir de ahí, se han realizado 2 ensayos clínicos: el estudio STRIVE realizado en 144 pacientes de 12 o más años36 y el estudio ENVISION realizado en 52 niños de entre 6 y 11 años37. Ambos estudios eran pacientes con al menos una mutación Gly551Asp, FEV1 entre 40-105%, seguidos durante un periodo de 14 días para posteriormente aleatorizarse a recibir 150mg de ivacaftor 2 veces al día vía oral o placebo durante un periodo de estudio de 48 semanas. Tras completar las 48 semanas de tratamiento se les ofreció a los pacientes continuar en un estudio abierto longitudinal, estudio PERSIST (NCT01117012)38, durante 96 semanas.
En el estudio STRIVE los pacientes del grupo de ivacaftor presentaron una mejoría del 10,6% en el FEV1 (objetivo primario del estudio) desde el día 15 del tratamiento que se mantuvo durante las 48 semanas del estudio. Además, se observó una disminución de la concentración de cloro en sudor (-48,7mmol/L de media), una mejoría de la calidad de vida, una reducción del 55% de las exacerbaciones y un aumento del peso de 2,7 kg.
Los resultados del estudio ENVISION son muy superponibles al estudio de los adolescentes y adultos, con la diferencia de que la calidad de vida no alcanzó la diferencia estadística. Tanto en el estudio STRIVE como en el ENVISION los efectos adversos encontrados más frecuentemente en el grupo de tratamiento fueron infecciones del tracto respiratorio superior, congestión nasal, dolor de garganta, mareos y exantema. Los resultados preliminares de las 12 primeras semanas del estudio PERSIST revelan que las mejoras en la función pulmonar (FEV1), los síntomas respiratorios y el aumento de peso entre pacientes que han sido tratados con ivacaftor se mantienen en este tiempo. Además en el subgrupo de pacientes que pasaron de placebo a ivacaftor al inicio del estudio PERSIST mostraron mejorías del FEV1 del 10,8% a los 15 días y del 13% a las 12 semanas, junto con una reducción de las exacerbaciones39.
Tras los buenos resultados de ivacaftor en niños mayores de 6 años y adultos sobre la mutación Gly551Asp a las 48 semanas, algunas cuestiones quedan pendientes por resolver. En primer lugar, el fármaco no se ha probado en niños menores de 6 años. Sin embargo, teniendo en cuenta que la afectación pulmonar comienza antes de los 6 años, tiene sentido poder corregir el defecto antes de que se produzca un daño irreversible. El ensayo clínico en este rango de edad está actualmente en marcha (NCT01705145)40. En segundo lugar, otra alternativa sería también probarlo en otras mutaciones de la clase iii. En este sentido, estudios in vitro sobre otras 9 mutaciones han demostrado unos resultados muy similares41, por lo que es razonable esperar que los resultados en pacientes sean también similares. Actualmente está en marcha un ensayo clínico en fase iii para pacientes mayores de 6 años con otras mutaciones de clase iii (estudios KONTINUE y KONNECTION; NCT01614470)42,43. En tercer lugar, aunque no existen tratamientos para el resto de las clases de mutaciones (iv a vi), los potenciadores del CFTR podrían ser igualmente beneficiosos en estos casos. En este sentido, actualmente se está llevando a cabo un ensayo clínico para evaluar la eficacia de ivacaftor en la mutación Arg117His de clase iv (estudio KONDUCT; NCT01614457)44. Finalmente, su eficacia y seguridad a largo plazo más allá de las 48 semanas aún no han sido dilucidadas. El G551D Observational Study (GOAL; NCT01521338)45 es un estudio observacional de seguimiento de pacientes mayores de 6 años en tratamiento con ivacaftor que informará sobre la eficacia y seguridad de ivacaftor a largo plazo con otros resultados de interés que incluyen mediadores inflamatorios en el esputo, aclaramiento mucociliar y pH gastrointestinal. Los resultados se esperan para finales de 2013.
ConclusionesLa FQ es una enfermedad autosómica recesiva monogénica que constituye un ejemplo de lo que podría ser una enfermedad bien situada para poder beneficiarse de la medicina personalizada. En la actualidad, 2 enfoques tienen por objetivo corregir el defecto básico: la terapia génica, dirigida a corregir la alteración genética, y la terapia con moléculas encaminadas a corregir el defecto funcional a nivel de la proteína. Esta última comienza a dar resultados prometedores con diversas moléculas en desarrollo y una de ellas (ivacaftor) ya comercializada para la mutación de clase iii Gly551Asp, con excelentes resultados en niños mayores de de 6 años, adolescentes y adultos. El objetivo final será a través de la terapia correctora y potenciadora poder llegar a dar respuesta a todos los pacientes con FQ cualquiera que sea su mutación. Es probable que, conforme se vayan teniendo resultados de estas y otras moléculas nuevas, haga falta una molécula o combinación de estas según la o las mutaciones existentes en cada paciente, pero el futuro es prometedor y se están dando importantes pasos para conseguir un tratamiento que consiga actuar de forma efectiva sobre la causa de la enfermedad.
Conflicto de interesesLas autoras declaran no tener ningún conflicto de intereses.







