


El diagnóstico de la afectación metastásica ganglionar en el cáncer de pulmón constituye un problema, a pesar de los avances en la estadificación. La determinación del estado de metilación en ganglios podría mejorar la capacidad de las técnicas citohistológicas para detectar afectación metastásica. Nuestro objetivo fue demostrar la viabilidad de realizar estudios de metilación en muestras ganglionares citológicas.
MétodosEstudio prospectivo que incluyó 88 pacientes con diagnóstico o alta sospecha de cáncer de pulmón no microcítico, en los que se realizó una punción citológica por ecobroncoscopia de adenopatías mediastínicas y/o hiliares. Se extrajo ADN a partir de muestras citológicas ganglionares y se realizó el tratamiento con bisulfito de sodio. Los estudios de metilación se realizaron por qPCR-MS y pirosecuenciación en los genes p16/INK4a y SHOX2.
ResultadosLa metodología empleada permitió obtener ADN de características óptimas/buenas en el 90% de los casos. No se observaron diferencias en la concentración de ADN respecto a la estación ganglionar ni al diagnóstico final. Los análisis por qPCR-MS y pirosecuenciación no fueron posibles en un reducido número de muestras debido a baja concentración de ADN, además de la inadecuada pureza, fragmentación y/o degradación debido al tratamiento con bisulfito de sodio.
ConclusiónLa cuantificación de la metilación por técnicas como qPCR-MS o pirosecuenciación en muestras ganglionares obtenidas por ecobroncoscopia resulta viable siempre y cuando se logre obtener una concentración adecuada de ADN, contribuyendo a la búsqueda de biomarcadores epigenéticos que mejoren la toma de decisiones en el cáncer de pulmón potencialmente curable en beneficio del paciente.
The diagnosis of microscopic lymph node metastasis in lung cancer is challenging despite the constant advances in tumor staging. The analysis of the methylation status of certain genes in lymph node samples could improve the diagnostic capability of conventional cyto-histological methods. The aim of this study was to demonstrate the feasibility of methylation studies using cytological lymph node samples.
MethodsProspective study including 88 patients with a diagnosis or strong suspicion of non-small cell lung cancer, in which an echobronchoscopy was performed on mediastinal or hilar lymph nodes for diagnostic and/or staging. DNA was extracted from cytological lymph node samples and sodium bisulfite modification was performed. Methylation studies for p16/INK4a and SHOX2 were accomplished by MS-qPCR and pyrosequencing.
ResultsThe methodology used in our study yielded optimal/good DNA quality in 90% of the cases. No differences in DNA concentration were observed with respect to the lymph node biopsied and final diagnosis. Methylation analyses using MS-qPCR and pyrosequencing were not possible in a small number of samples mainly due to low DNA concentration, inadequate purity, fragmentation and/or degradation as a consequence of bisulfite conversion.
ConclusionMethylation quantification using MS-qPCR and pyrosequencing of cytological lymph node samples obtained using echobronchoscopy is feasible if an appropriate DNA concentration is obtained, notably contributing to the identification of epigenetic biomarkers capable of improving decision-making for the benefit of potentially curable lung cancer patients.
El cáncer de pulmón (CP) es actualmente uno de los principales problemas de salud pública y la causa más frecuente de muerte por cáncer1. Ello se debe a que la mayoría de los pacientes comienzan en estadios avanzados de la enfermedad, donde las opciones de tratamientos curativos, fundamentalmente quirúrgicos, no existen. En estos últimos años se han producido importantes avances en el desarrollo de nuevas técnicas de imagen, como la tomografía por emisión de positrones-tomografía computarizada, de obtención de muestras a través de la ecoendoscopia endobronquial (USEB) o esofágica, y de exploración quirúrgica del mediastino con los nuevos sistemas con cámara de vídeo incorporada al mediastinoscopio (mediastínica y transcervical), así como la videotoracoscopia, resultando más rentables y mejorando la precisión de la estadificación2,3. Sin embargo, el 30-40% de los pacientes sin afectación ganglionar aparente en la cirugía sufrirán una recidiva tumoral4. El diagnóstico de la afectación ganglionar por el tumor plantea muchas dificultades y resulta el factor más importante en el pronóstico y tratamiento de pacientes con CP potencialmente resecable. Más aún, la afectación microscópica del ganglio linfático (N oculto) puede no ser evidenciada mediante las técnicas citohistológicas rutinarias de anatomía patológica, siendo necesaria una estadificación ganglionar más precisa3,5. La búsqueda de estrategias que permitan mejorar estos aspectos se hace esencial.
Los avances en el estudio de la epigenética, referidos especialmente a la metilación del ADN, han demostrado su potencial aplicabilidad en el CP6,7 para diagnóstico, pronóstico y respuesta al tratamiento; sin embargo, hasta el momento existen conocimientos limitados acerca de su potencial aplicabilidad en la estadificación mediastínica e hiliar. El objetivo de este trabajo fue demostrar la viabilidad para realizar estudios de metilación valorando la cantidad y calidad del ADN extraído a partir de muestras ganglionares citológicas procedentes de punción transbronquial (PTB) mediante USEB en pacientes con carcinoma pulmonar no microcítico.
MétodosSujetos de estudioEstudio prospectivo en el que se incluyeron pacientes consecutivos con diagnóstico o alta sospecha de carcinoma pulmonar no microcítio, en los que se realizó una punción citológica dirigida por USEB de adenopatías mediastínicas o hiliares con fin diagnóstico y/o de estadificación. La inclusión de los casos se realizó en nuestro comité multidisciplinar de CP siguiendo el protocolo de diagnóstico, estadificación y tratamiento del CP de nuestro centro (www.chuvi.es), que sigue las normativas y recomendaciones nacionales e internacionales actuales. Se incluyeron pacientes con adenopatías hiliares y/o mediastínicas de eje menor mayor de 1cm en la tomografía computarizada o de 5mm en caso de hipercaptación patológica en la tomografía por emisión de positrones-tomografía computarizada. Se excluyeron aquellos pacientes con muestra no valorable según el citólogo o con muestra insuficiente para estudios posteriores de metilación. Todos los procedimientos se realizaron en la Unidad de Técnicas Broncopleurales del Servicio de Neumología del CHUVI.
El estudio se llevó a cabo de acuerdo con los principios clínico-éticos del Gobierno de España y la Declaración de Helsinki, y fue aprobado por el Comité Ético de Investigación Clínica de Galicia. Se obtuvo el consentimiento informado de todos los pacientes y se garantizó el anonimato.
Procedimiento de ecoendoscopia endobronquial-punción transbronquialLos procedimientos se realizaron de forma consecutiva entre septiembre de 2009 y diciembre de 2012, siendo la mayoría ambulatorios, en la sala de broncoscopias convencional, con monitorización ecocardiográfica, tensional y pulsioximétrica, y bajo sedación consciente con midazolam y fentanilo. Se utilizó un broncoscopio BF-UC180F-OL8 (Olympus, Tokio, Japón) y un ecógrafo Aloka ProSound Alpha 5 (Hitachi-Aloka, Tokio, Japón). Tras la exploración endoscópica se procedió a la medición de la lesión y a su punción utilizando agujas NA2015X-4022 (Olympus, Tokio, Japón), avanzando por el canal del ecobroncoscopio hasta la vía aérea, donde la aguja era extraída e insertada en el interior del ganglio a través de la pared traqueal y bronquial con control ultrasonográfico, y movilizada al interior del ganglio realizándose aspiración a jeringa de vacío. Se realizaron de uno a 3 pases por adenopatía en función de los resultados inmediatos obtenidos. Se recuperó el material obtenido, la muestra se fijó en alcohol y fue examinada inmediatamente por un citólogo, quien la identificó como: procedente de un ganglio normal (celularidad predominantemente linfoide sin atipia y/o material antracótico); ganglio con infiltración neoplásica (presencia de células neoplásicas y celularidad de ganglio); muestra no valorable (presencia únicamente de células sanguíneas o bronquiales). Una parte de las muestras verificada in situ como representativa se resuspendió en solución salina estéril e inmediatamente se congeló a −20°C para los posteriores estudios moleculares.
En aquellos casos en los que el resultado anatomopatológico no evidenció afectación neoplásica se hizo una comprobación del resultado negativo mediante cirugía torácica. En el caso de los pacientes en los que se decidió no realizar una confirmación quirúrgica, se realizó un seguimiento mediante revisión de la historia clínica durante un año. Si la adenopatía no experimentó crecimiento durante ese año se consideró como verdadero negativo8. Todos los casos dudosos se excluyeron del estudio.
Extracción de ADN a partir de muestras ganglionaresEl ADN se extrajo utilizando el kit QIAamp DNA Mini Kit (Qiagen, Valencia, CA, EE. UU.), adaptando el protocolo al volumen de cada muestra. El ADN se eluyó con 50μL de agua y se hicieron 2 alícuotas para posteriores tratamientos con bisulfito de sodio. La concentración de ADN se midió en un NanoDrop 2000c (Thermo Scientific). Las ratios de absorbancia 260/280nm permitieron estimar su pureza como: óptima (ratio 1,7-2,0), buena (ratio>2,0), o presencia de proteínas u otros contaminantes (ratio<1,7). El ADN se almacenó a −20°C hasta su uso.
Tratamiento con bisulfito de sodioPara llevar a cabo el análisis de metilación fue necesario tratar el ADN con bisulfito de sodio. Para ello empleamos el EZ DNA Methylation-Direct Kit (Zymo Research, Irvine, CA, EE. UU.) siguiendo el protocolo recomendado por el fabricante, eluyendo con 20μL. El ADN modificado pasa a ser de cadena simple y rico en uracilo, resultando químicamente similar al ARN, por lo que su concentración se determinó como si se tratase de este ácido nucleico empleando el NanoDrop (Thermo Scientific). Las ratios 260/280nm permitieron estimar su pureza como: óptima (ratio 1,8-2,2), buena (ratio>2,2), o presencia de proteínas u otros contaminantes (ratio<1,8). El ADN tratado se almacenó a −80°C hasta el momento de su uso.
Análisis de metilación por reacción en cadena de la polimerasa en tiempo real específica para metilaciónEl análisis de metilación de los genes p16/INK4a y SHOX2 se realizó mediante PCR en tiempo real (qPCR) anidada específica para (MS) partiendo de 3μL de ADN tratado con bisulfito. En la primera ronda de amplificación se emplearon primers externos que amplificaron la isla CpG independientemente de su estado de metilación. Seguidamente se realizó una qPCR-MS diluyendo el producto de PCR y empleando sondas y cebadores específicos de metilación en un equipo StepOne (Applied Biosystems). La información más detallada de las secuencias utilizadas está disponible a petición del lector.
Análisis de metilación por pirosecuenciaciónLa pirosecuenciación se efectuó para los genes p16/INK4a y SHOX2. Se realizó una PCR con 3μL de ADN modificado utilizando cebadores diseñados con el software PyroMark Assay Design v2.0 (Qiagen). Seguidamente se realizó la secuenciación y cuantificación de la metilación empleando el cebador de pirosecuenciación en un PyroMark Q24 (Qiagen). Las islas CpG estudiadasfueron las mismas que se estudiaron por qPCR-MS. La información más detallada de las secuencias utilizadas está disponible a petición del lector.
Análisis estadísticosPara los análisis estadísticos se utilizó el paquete SPSS (v19.0). Se emplearon pruebas bilaterales, y valores de p<0,05 se consideraron significativos. La concentración de ADN se expresó como mediana y rango intercuartil (RIC). La distribución normal y la homogeneidad de varianza se comprobaron con las pruebas de Kolmogorov-Smirnov y Levene, respectivamente. Los análisis no paramétricos para 2 muestras independientes se realizaron con la U de Mann-Whitney, mientras que para más de 2 muestras independientes se empleó la prueba de Kruskal-Wallis. Para analizar diferencias en datos pareados se utilizó la prueba de Wilcoxon. Las variables que se estudiaron fueron sexo, tipo de tumor, localización de las adenopatías, diagnóstico de la adenopatía, aspecto de la muestra ganglionar y concentración y pureza del ADN sin modificar y modificado con bisulfito de sodio.
La estimación del tamaño muestral necesario se ha realizado utilizando el programa Granmo 5.2 (Institut Municipal d’Investigació Médica, 1999), para muestras pareadas, aceptando un riesgo alfa de 0,05 y un riesgo beta de 0,20, asumiendo una prevalencia estimada de adenopatías malignas de un 70% y una diferencia mínima entre la sensibilidad y el valor predictivo negativo de las técnicas citohistológicas y las determinaciones moleculares del 15 y 10%, respectivamente. Se estimó que se precisan un mínimo de 79 casos para llevar a cabo el estudio.
ResultadosCaracterísticas de los pacientes y de las muestras citológicas ganglionaresSe incluyeron 88 pacientes, obteniéndose 170 muestras de estaciones ganglionares distintas por USEB-PTB, distribuidos de la siguiente manera: 38 pacientes con una muestra, 29 con 2 muestras, 13 con 3 muestras, 5 con 4 muestras, y 3 con 5 muestras. Setenta y cuatro pacientes (84%) eran varones, con una mediana de edad de 62,2 años (RIC: 51-73 años). En base a una escala colorimétrica propia, un 44,7% (n=76) de las muestras citológicas ganglionares mostró apariencia turbio-claro, un 38,2% (n=65) resultó poco sanguinolenta, y un 17,1% (n=29) fue muy sanguinolenta.
En un 86,5% de los casos (n=147) las punciones se realizaron en ganglios mediastínicos, mientras que el 13,5% (n=23) restante se realizó en ganglios hiliares. Las adenopatías más frecuentemente puncionadas fueron la 4R (39,4%; n=67) y la 7 (33,5%; n=57), seguida de la 4L (9,4%; n=16), 11L (7,1%; n=12), 11R (4,7%; n=8), 8 (2,4%; n=4), 2R (1,8%; n=3), 10R (1,2%; n=2) y 12R (0,6%; n=1). De acuerdo con el diagnóstico definitivo del ganglio, un 48,2% (n=82) fue benigno (no metastásico), mientras que el 51,8% (n=88) restante fue maligno (metastásico). Entre estos últimos, un 61,4% (n=54) fueron adenocarcinoma, un 31,8% (n=28) epidermoide y un 6,8% (n=6) de célula grande.
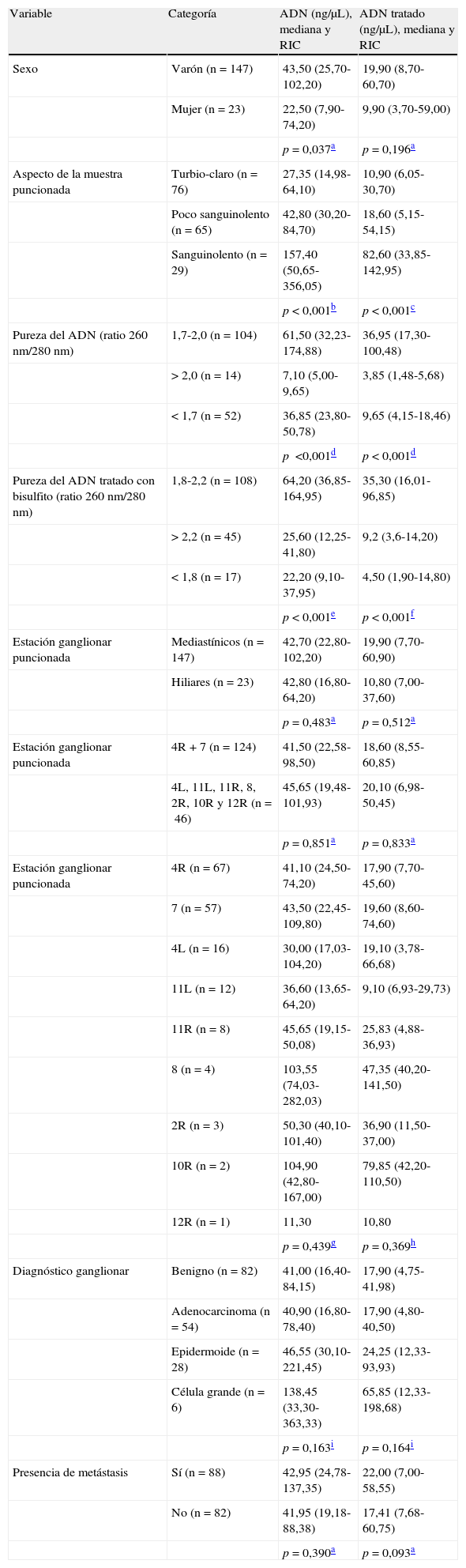
Características del ácido desoxirribonucleico extraído a partir de las muestras citológicas ganglionaresSe obtuvo un amplio rango de concentración de ADN de 4,10-1.103,00ng/μL (mediana: 42,75ng/μL; RIC: 22,48-100,13ng/μL). Para estudiar las características del ADN relacionamos su concentración con parámetros epidemiológicos y clínicos(tabla 1). Observamos que los varones muestran una mediana mayor, con diferencias significativas respecto a las mujeres (p=0,037). La concentración de ADN mostró diferencias en función del aspecto de las muestras citológicas, con valores mayores para muestras sanguinolentas, seguidas de las poco sanguinolentas, y por último, las de apariencia turbio-claro (p<0,001), conforme al contenido celular.
Características de las muestras ganglionares en función de la concentración de ADN extraído y tras el tratamiento con bisulfito de sodio
| Variable | Categoría | ADN (ng/μL), mediana y RIC | ADN tratado (ng/μL), mediana y RIC |
| Sexo | Varón (n=147) | 43,50 (25,70-102,20) | 19,90 (8,70-60,70) |
| Mujer (n=23) | 22,50 (7,90-74,20) | 9,90 (3,70-59,00) | |
| p=0,037a | p=0,196a | ||
| Aspecto de la muestra puncionada | Turbio-claro (n=76) | 27,35 (14,98-64,10) | 10,90 (6,05-30,70) |
| Poco sanguinolento (n=65) | 42,80 (30,20-84,70) | 18,60 (5,15-54,15) | |
| Sanguinolento (n=29) | 157,40 (50,65-356,05) | 82,60 (33,85-142,95) | |
| p<0,001b | p<0,001c | ||
| Pureza del ADN (ratio 260nm/280nm) | 1,7-2,0 (n=104) | 61,50 (32,23-174,88) | 36,95 (17,30-100,48) |
| >2,0 (n=14) | 7,10 (5,00-9,65) | 3,85 (1,48-5,68) | |
| <1,7 (n=52) | 36,85 (23,80-50,78) | 9,65 (4,15-18,46) | |
| p<0,001d | p<0,001d | ||
| Pureza del ADN tratado con bisulfito (ratio 260nm/280nm) | 1,8-2,2 (n=108) | 64,20 (36,85-164,95) | 35,30 (16,01-96,85) |
| >2,2 (n=45) | 25,60 (12,25-41,80) | 9,2 (3,6-14,20) | |
| <1,8 (n=17) | 22,20 (9,10-37,95) | 4,50 (1,90-14,80) | |
| p<0,001e | p<0,001f | ||
| Estación ganglionar puncionada | Mediastínicos (n=147) | 42,70 (22,80-102,20) | 19,90 (7,70-60,90) |
| Hiliares (n=23) | 42,80 (16,80-64,20) | 10,80 (7,00-37,60) | |
| p=0,483a | p=0,512a | ||
| Estación ganglionar puncionada | 4R+7 (n=124) | 41,50 (22,58-98,50) | 18,60 (8,55-60,85) |
| 4L, 11L, 11R, 8, 2R, 10R y 12R (n=46) | 45,65 (19,48-101,93) | 20,10 (6,98-50,45) | |
| p=0,851a | p=0,833a | ||
| Estación ganglionar puncionada | 4R (n=67) | 41,10 (24,50-74,20) | 17,90 (7,70-45,60) |
| 7 (n=57) | 43,50 (22,45-109,80) | 19,60 (8,60-74,60) | |
| 4L (n=16) | 30,00 (17,03-104,20) | 19,10 (3,78-66,68) | |
| 11L (n=12) | 36,60 (13,65-64,20) | 9,10 (6,93-29,73) | |
| 11R (n=8) | 45,65 (19,15-50,08) | 25,83 (4,88-36,93) | |
| 8 (n=4) | 103,55 (74,03-282,03) | 47,35 (40,20-141,50) | |
| 2R (n=3) | 50,30 (40,10-101,40) | 36,90 (11,50-37,00) | |
| 10R (n=2) | 104,90 (42,80-167,00) | 79,85 (42,20-110,50) | |
| 12R (n=1) | 11,30 | 10,80 | |
| p=0,439g | p=0,369h | ||
| Diagnóstico ganglionar | Benigno (n=82) | 41,00 (16,40-84,15) | 17,90 (4,75-41,98) |
| Adenocarcinoma (n=54) | 40,90 (16,80-78,40) | 17,90 (4,80-40,50) | |
| Epidermoide (n=28) | 46,55 (30,10-221,45) | 24,25 (12,33-93,93) | |
| Célula grande (n=6) | 138,45 (33,30-363,33) | 65,85 (12,33-198,68) | |
| p=0,163i | p=0,164i | ||
| Presencia de metástasis | Sí (n=88) | 42,95 (24,78-137,35) | 22,00 (7,00-58,55) |
| No (n=82) | 41,95 (19,18-88,38) | 17,41 (7,68-60,75) | |
| p=0,390a | p=0,093a |
RIC: rango intercuartil.
En la tabla se muestran los valores p obtenidos con el test de Kruskal-Wallis. Como prueba post-hoc del Kruskal-Wallis se utilizó la prueba de U de Mann-Whitney comparando por parejas las categorías de cada variable.
El protocolo empleado permitió extraer ADN de pureza óptima o buena (ratio 260/280nm) en un 61,18 y 8,24% de los casos, respectivamente. Las mayores concentraciones de ADN correspondieron a muestras de pureza óptima, seguidas de aquellas con contaminantes,y por último, las de pureza buena (p<0,001; tabla 1).
En relación con la estación ganglionar puncionada, no observamos diferencias en la concentración de ADN al comparar ganglios mediastínicos con hiliares (p=0,483), ni entre los ganglios mayoritarios 4R y 7 en relación con el resto (p=0,851). No se observaron diferencias en la concentración de ADN entre todas las adenopatías desglosadas en función de su localización. Sin embargo, las adenopatías 8 presentaron una mayor concentración de ADN en relación con las estaciones 4R, 11R y 11L (tabla 1). Para los grupos diagnósticos la concentración de ADN no siguió una distribución normal ni presentó homogeneidad de varianza, observándose medianas alrededor de 40ng/μL para las categorías benigno, adenocarcinoma y epidermoide, y de 138,4ng/μL para los 6 casos de célula grande (tabla 1). Consecuentemente, no se detectaron diferencias en la concentración de ADN en función del diagnóstico ganglionar (p=0,163)o entre adenopatías benignas y malignas (p=0,390).
Características del ácido desoxirribonucleico tras el tratamiento con bisulfito de sodioSimilarmente, se estudió la concentración de ADN modificado en relación con algunas variables(tabla 1). Se registraron concentraciones menores a las previas al tratamiento, con un rango de 0,20-392,30ng/μL (mediana: 19,13ng/μL; RIC: 7,60-60,18ng/μL). Al contrastar las concentraciones antes y después del tratamiento observamos diferencias significativas (p<0,001), con concentraciones disminuidas en el 93,53% de las muestras.
Aunque los varones siguieron mostrando mayores concentraciones de ADN que las mujeres, las diferencias no resultaron significativas (p=0,196). En relación con el aspecto de las muestras, las diferencias en la concentración de ADN modificado resultaron significativas (p<0,001). Por otra parte, la estimación de la pureza del ADN modificado indicó que el 63,53 y 26,47% son de calidad óptima o buena, respectivamente, totalizando un 90% los potencialmente adecuados para realizar el análisis de metilación. La mayor mediana correspondió a muestras de pureza óptima, al menos 3 veces superior a las muestras de purezas inferiores (p<0,001).
En relación con la estación ganglionar puncionada, como se observa en la tabla 1 no se detectaron diferencias entre las adenopatías mediastínicas e hiliares (p=0,512) ni entre las estaciones 4R y 7 respecto al resto (p=0,833). De manera similar a lo observado con la concentración de ADN antes del tratamiento, las diferencias observadas en el ADN modificado se presentaron entre las adenopatías 2R, 4R, 11R y 11L respecto a las 8 (tabla 1). Por último, no se encontraron diferencias al comparar las categorías de diagnóstico ganglionar (p=0,164) ni las adenopatías benignas y malignas (p=0,093) (tabla 1).
Estudios de metilación a partir de muestras citológicas ganglionaresAunque la mayoría de las muestras parecen adecuadas para estudios posteriores, para corroborar su validez se realizaron análisis de metilación para 2 genes supresores de tumor empleando 2 metodologías distintas. En la tabla 2 se resumen las características de las muestras en función de la obtención del dato de metilación (muestras con/sin dato de metilación), para ambos genes y los 2 abordajes.
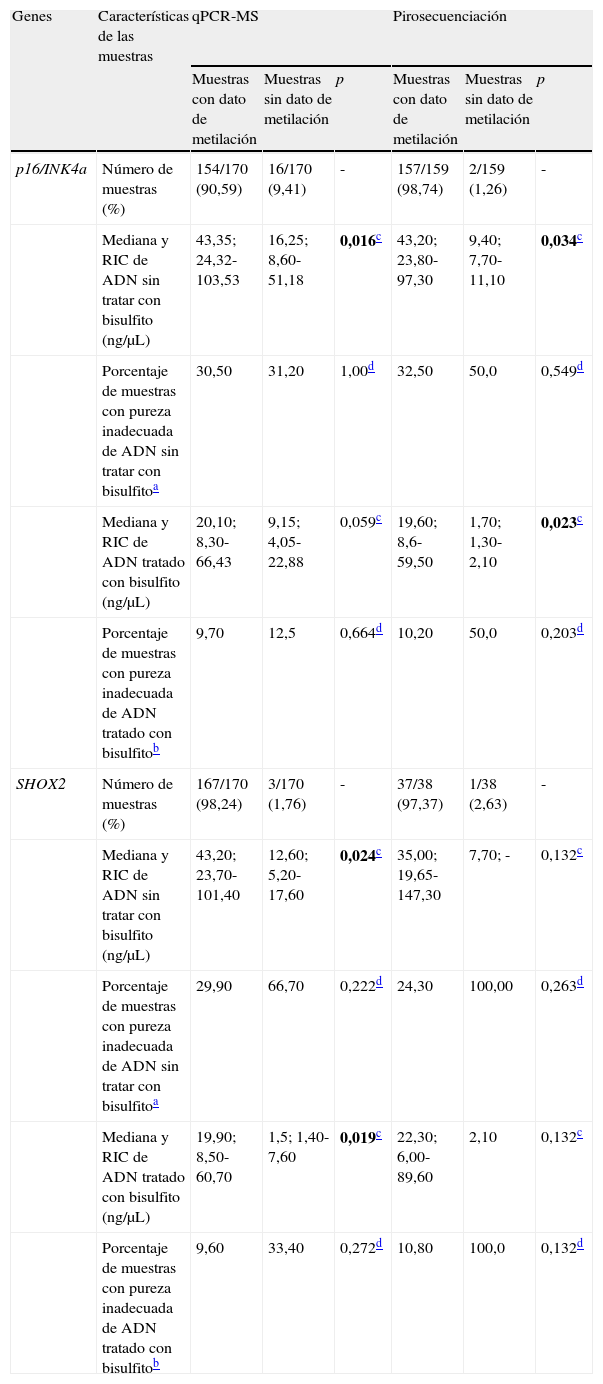
Características de las muestras ganglionares para los análisis de metilación de p16/INK4a y SHOX2
| Genes | Características de las muestras | qPCR-MS | Pirosecuenciación | ||||
| Muestras con dato de metilación | Muestras sin dato de metilación | p | Muestras con dato de metilación | Muestras sin dato de metilación | p | ||
| p16/INK4a | Número de muestras (%) | 154/170 (90,59) | 16/170 (9,41) | - | 157/159 (98,74) | 2/159 (1,26) | - |
| Mediana y RIC de ADN sin tratar con bisulfito (ng/μL) | 43,35; 24,32-103,53 | 16,25; 8,60-51,18 | 0,016c | 43,20; 23,80-97,30 | 9,40; 7,70-11,10 | 0,034c | |
| Porcentaje de muestras con pureza inadecuada de ADN sin tratar con bisulfitoa | 30,50 | 31,20 | 1,00d | 32,50 | 50,0 | 0,549d | |
| Mediana y RIC de ADN tratado con bisulfito (ng/μL) | 20,10; 8,30-66,43 | 9,15; 4,05-22,88 | 0,059c | 19,60; 8,6-59,50 | 1,70; 1,30-2,10 | 0,023c | |
| Porcentaje de muestras con pureza inadecuada de ADN tratado con bisulfitob | 9,70 | 12,5 | 0,664d | 10,20 | 50,0 | 0,203d | |
| SHOX2 | Número de muestras (%) | 167/170 (98,24) | 3/170 (1,76) | - | 37/38 (97,37) | 1/38 (2,63) | - |
| Mediana y RIC de ADN sin tratar con bisulfito (ng/μL) | 43,20; 23,70-101,40 | 12,60; 5,20-17,60 | 0,024c | 35,00; 19,65-147,30 | 7,70; - | 0,132c | |
| Porcentaje de muestras con pureza inadecuada de ADN sin tratar con bisulfitoa | 29,90 | 66,70 | 0,222d | 24,30 | 100,00 | 0,263d | |
| Mediana y RIC de ADN tratado con bisulfito (ng/μL) | 19,90; 8,50-60,70 | 1,5; 1,40-7,60 | 0,019c | 22,30; 6,00-89,60 | 2,10 | 0,132c | |
| Porcentaje de muestras con pureza inadecuada de ADN tratado con bisulfitob | 9,60 | 33,40 | 0,272d | 10,80 | 100,0 | 0,132d | |
RIC: rango intercuartil.
En negrita, el valor de la p significativo.
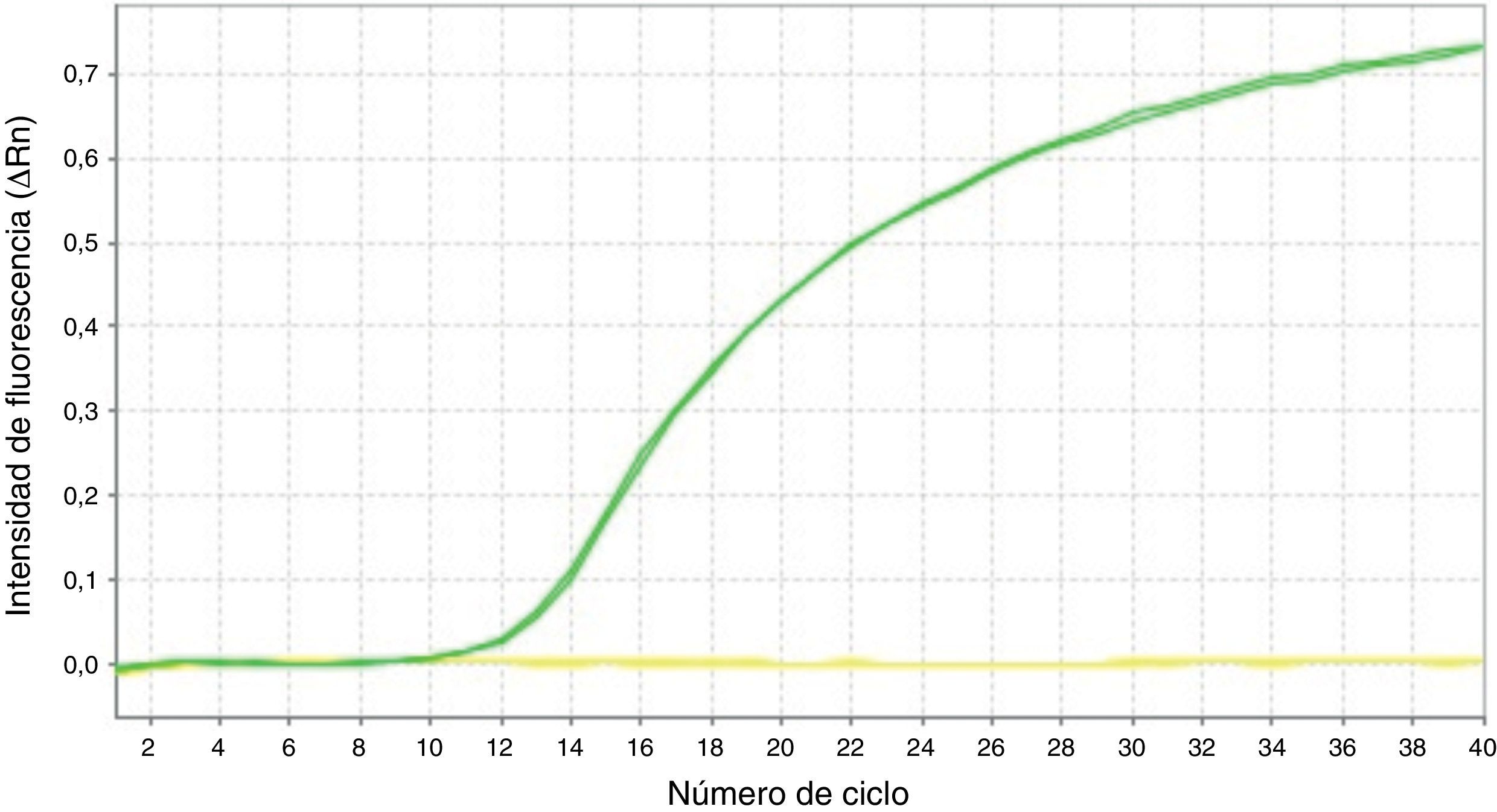
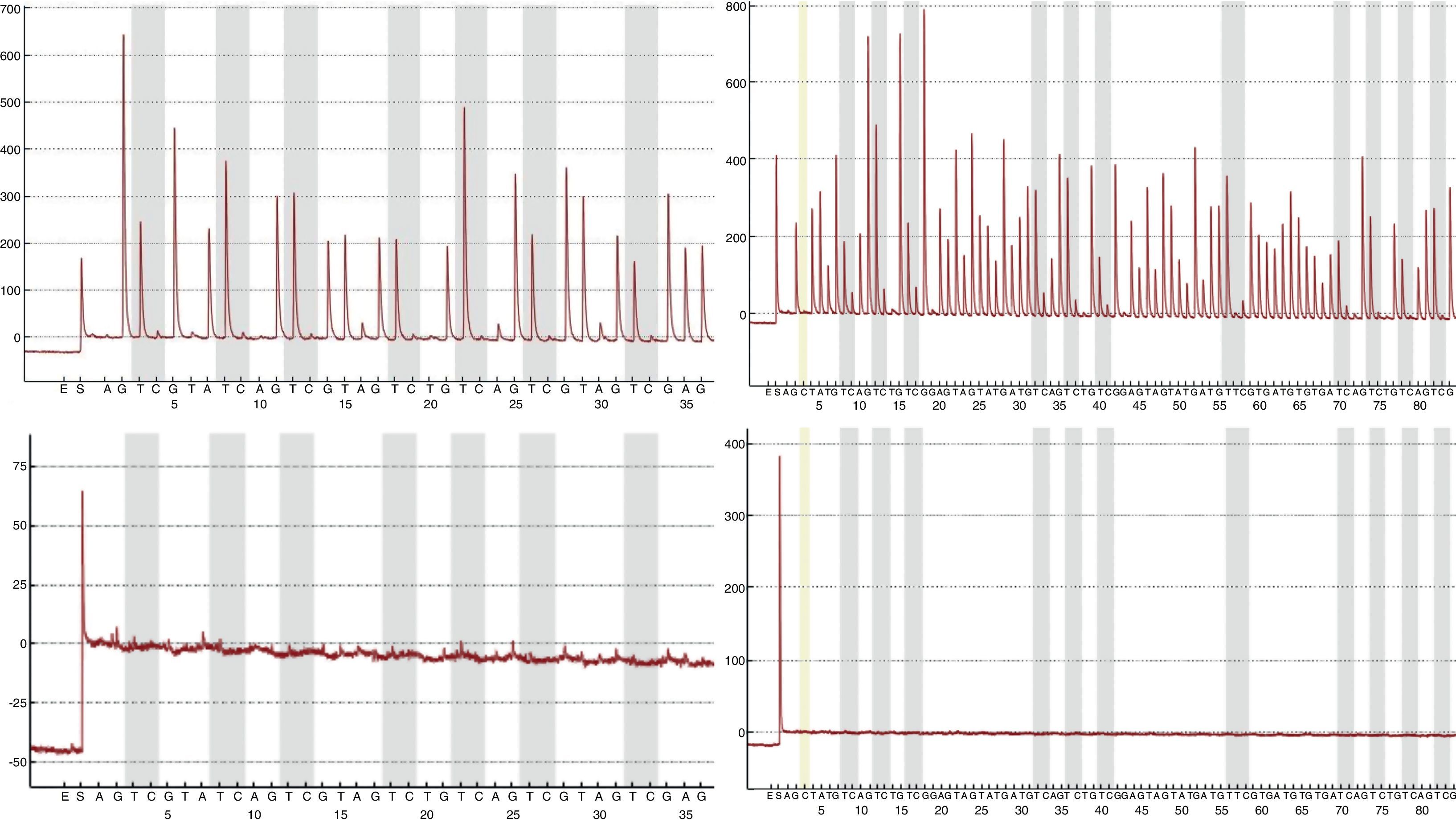
Mediante la técnica de qPCR-MS no fueron posibles los análisis en el 9,41 (16/170; p16/INK4a) y 1,76% (3/170; SHOX2) de las muestras, mientras que por pirosecuenciación no se obtuvo información en un 1,26 (2/159; p16/INK4a) y 2,63% (1/38; SHOX2) de los casos. Las medianas y los RIC de la concentración de ADN antes y después del tratamiento resultaron considerablemente inferiores en las muestras sin dato de metilación en comparación con aquellas en las que sí fue posible el análisis. Esta diferencia resultó estadísticamente significativa (tabla 2), relacionándose con la dificultad en los análisis para ciertas muestras. Sin embargo, el porcentaje de muestras con pureza inadecuada se mostró muy variable tanto para las muestras sin dato de metilación como para aquellas con dato, aunque en las primeras resultó en todos los casos mayor, relacionándose también con la ausencia de resultados con las técnicas empleadas. En la figura 1 se muestran las curvas de amplificación de p16/INK4a mediante qPCR para una muestra válida y una no válida, mientras que los pirogramas de p16/INK4a para una muestra válida y una no válida se presentan en la figura 2.
 y una muestra no válida (línea amarilla).")
 y una muestra no válida (gráficas inferiores). Como la pirosecuenciación se realizó en 2 reacciones distintas para abarcar los 18 sitios CpG de la isla, los pirogramas de la izquierda muestran los primeros 7 sitios CpG, mientras que los pirogramas de la derecha muestran los últimos 11 sitios CpG. Las barras grises de los pirogramas indican cada sitio CpG.")
Pirogramas de p16/INK4a para una muestra válida (gráficas superiores) y una muestra no válida (gráficas inferiores). Como la pirosecuenciación se realizó en 2 reacciones distintas para abarcar los 18 sitios CpG de la isla, los pirogramas de la izquierda muestran los primeros 7 sitios CpG, mientras que los pirogramas de la derecha muestran los últimos 11 sitios CpG. Las barras grises de los pirogramas indican cada sitio CpG.
Existe amplia evidencia de la presencia de micrometástasis oculta en ganglios, indetectable por métodos citohistológicos estándares5,9–12. La reciente introducción de técnicas moleculares ha demostrado una mejora en la detección de células metastásicas tanto en tejidos, incluyendo el material citológico, como en diferentes fluidos biológicos9. Recientemente, Nakajima et al.11 describieron que el aislamiento simultáneo de ADN, ARN y proteína puede realizarse a partir de muestras obtenidas por USEB. Sin embargo, hasta la fecha son pocos los datos publicados sobre análisis moleculares empleando estas muestras5,10,11, incluidos estudios de expresión por qPCR de citoqueratinas, MUC1, p53, K-ras, CEA, CK19, KS1/4, lunx, PDEF y ALK13–15, detección de mutaciones en EGFR, KRAS, BRAF y PIK3CA16, y detección de citoqueratinas y ALK por inmunohistoquímica13,15,17. Asimismo, dada la importancia de la metilación aberrante de islas CpG como principal mecanismo epigenético para la regulación del silenciamiento de los genes y su implicación en los procesos neoplásicos, los estudios moleculares también se han centrado en esta área, estudiándose el estado de metilación en los genes MGMT, p16/INK4a, p14/ARF, FANCF, Reprimo, TMS1/ASC y AP-2α9,18.
En este trabajo hemos demostrado que las muestras ganglionares obtenidas por USEB son adecuadas para realizar estudios de metilación empleando técnicas de qPCR-MS y pirosecuenciación, con mayor sensibilidad que la PCR-MS comúnmente empleada en los análisis de metilación, incluidos los estudios referenciados previamente9,18.
A diferencia de otros estudios moleculares con muestras de sangre, el inconveniente inicial de emplear muestras obtenidas por USEB es el volumen limitante recuperado en cada punción, además de la variación en la celularidad, por lo que es prioritario extraer la mayor cantidad de ADN de buena calidad. El protocolo empleado en nuestro estudio permitió obtener ADN de pureza óptima/buena en un 69% de las muestras. Además, no se observaron variaciones según la estación ganglionar puncionada ni el diagnóstico histológico final de la neoplasia.
La metilación del ADN, a diferencia de las mutaciones acompañadas de cambios en la secuencia de ADN, consiste en la transferencia de grupos metilos (-CH3) a bases citosina del ADN situadas previa y contiguamente a una guanina. Por ello, un paso previo para el estudio de metilación constituye el tratamiento con bisulfito de sodio, que convierte todas las citosinas no metiladas en uracilos, mientras que las citocinas metiladas permanecen inalteradas19,20. Esta modificación resulta indispensable para poder distinguir las citosinas metiladas de las no metiladas.
Durante el tratamiento con bisulfito el ADN es sometido a condiciones agresivas (largas incubaciones a altas temperaturas y pH ácido, altas molaridades de bisulfito y urea) para asegurar la completa conversión20, siendo un punto crítico para realizar un correcto análisis de metilación evitando la sobrestimación21. El kit que empleamos para el tratamiento de bisulfito está diseñado para minimizar la degradación y pérdida del ADN. La cantidad óptima de ADN de partida es de 200-500ng (10-25ng/μL), y solo el 8,82% de las muestras del estudio presentaron concentraciones inferiores, encontrándose aún así dentro del amplio rango (50pg-2μg) recomendado.
Aunque se estima que el kit que empleamos permite una recuperación del ADN inicial mayor del 80%, un 93,53% de las muestras tratadas mostraron menor concentración, con una media de disminución del 52,92±38,77%. A pesar de esta disminución, la calidad del ADN modificado resultó óptima/buena en el 90% de las muestras. Ehrich et al. (2007)20, empleando el mismo kit de bisulfito que utilizamos en este estudio, diseñaron un ensayo para determinar la calidad del ADN modificado y observaron que se consigue buena calidad de amplificación de fragmentos de hasta 477pb, disminuyendo su calidad en los de 617pb.
Si bien la cantidad y calidad del ADN modificado a priori parece adecuada para estudios posteriores, la validez de las muestras se determinó empleando las técnicas de qPCR-MS y pirosecuenciación. La qPCR-MS es un método cuantitativo, obteniéndose un porcentaje de metilación relativo de toda la isla CpG, mientras que la pirosecuenciación se emplea como gold estandar en los análisis de metilación, obteniéndose el porcentaje de metilación para cada sitio CpG de la isla estudiada.
Los genes seleccionados fueron p16/INK4a y SHOX2, descritos ampliamente en la literatura como potenciales biomarcadores de metilación para el pronóstico y diagnóstico del CP22–25. El gen p16/INK4a está implicado en la regulación del ciclo celular induciendo su interrupción, mientras que el gen SHOX2 pertenece a la familia de genes con dominio homeobox y está relacionado con el empaquetamiento del ADN. La metilación de estos genes ha sido estudiada por PCR-MS, qPCR-MS, pirosecuenciación y enzimas de restricción sensibles a sitios de metilación, entre otros, empleando muestras de tumor primario, suero, plasma, esputo, aspirado o lavado bronquial22,23,25.
La isla CpG que estudiamos en p16/INK4a es de 150pb e incluye 18 sitios CpG, mientras que para SHOX2 esta corresponde a 124pb con 11 sitios CpG, cercanos al rango 150-300pb recomendado para obtener buena calidad de amplificación20,21. La cuantificación de la metilación no fue posible en algunas muestras debido a la baja cantidad de ADN, además de la inadecuada pureza, fragmentación y degradación como consecuencia del tratamiento, justificando la dificultad de los análisis.
La concentración de ADN entre 2,6-4,6 veces mayor en las muestras con dato de metilación en relación con aquellas sin dato pone en evidencia la importancia de la cantidad de ADN de partida. En cuanto a la concentración de ADN luego del tratamiento de bisulfito, las diferencias resultaron aún mayores, entre 2,2-13 veces superiores. De acuerdo con nuestro estudio, es conveniente que a partir de muestras obtenidas por USEB se logre conseguir una concentración de ADN de al menos 20ng/μL para ser tratado con bisulfito de sodio. Asimismo, para lograr cuantificar la metilación mediante qPCR-MS anidada o pirosecuenciación es recomendable una concentración de ADN tratado no inferior a 8ng/μL.
Resulta razonable comentar que en los análisis por qPCR-MS la ausencia de metilación de p16/INK4a probablemente explique la mayor cantidad de muestras sin datos, ya que por pirosecuenciación los análisis fueron posibles en prácticamente todas las muestras. Para el gen SHOX2, aunque el 2,63% de las muestras no fueron posibles de pirosecuenciar, cabe comentar que tan solo se pirosecuenciaron 38 muestras, resultando sin dato una única muestra que tampoco pudo ser pirosecuenciada para p16/INK4a.
Los avances en las técnicas menos invasivas para la obtención de muestras como la USEB, junto con el mejor entendimiento de las bases moleculares del desarrollo y progresión del tumor como la metilación aberrante del ADN, constituyen herramientas que en conjunto representan una estrategia muy prometedora para la mejora en el diagnóstico y estadificación del CP. Para trasladar esta estrategia a la práctica clínica resulta imprescindible un adecuado material de partida en términos de cantidad y calidad del ADN. De acuerdo con el estudio que realizamos, el abordaje multidisciplinar es posible, resultando, además, de gran rendimiento, ya que permitiría el análisis por qPCR-MS (PCR anidada) de hasta 12 marcadores de metilación. Más aún, la posibilidad de tener suficiente muestra en condiciones nos permitirá la combinación de un panel de marcadores que mejoraría sin duda la sensibilidad requerida para la detección de la afectación microscópica del ganglio linfático (N oculto), no conseguida con las actuales técnicas citohistológicas.
ConclusiónLa cuantificación de la metilación por técnicas como qPCR-MS o pirosecuenciación a partir de muestras ganglionares obtenidas por USEB-PTB resulta viable siempre y cuando se logre obtener una concentración mínimamente adecuada de ADN. La posibilidad de realizar estudios de epigenética en muetras ganglionares obtenidas por USEB podría contribuir notablemente a la búsqueda de biomarcadores epigenéticos que mejoren la toma de decisiones en el CP potencialmente curable en beneficio del paciente.
FinanciaciónPrograma Sectorial de Investigación Aplicada (INCITE, Consellería de Innovación e Industria de la Xunta de Galicia). Código 09CSA053905PR.
Fondos de Investigación Sanitaria del Instituto Carlos III. Código PI09/90385.
Las muestras empleadas pertenecen al biobanco del CHUVI (RETIC-FIS-ISCIII RD09/0076/00011).
E. U. Seventh Framework Program (FP7/REGPOT-2012-2013-1. Code: N.o 316265, BIOCAPS.
AutoríaConcepto del estudio y diseño: Leiro-Fernández, De Chiara, Fernández-Villar, Botana-Rial.
Inclusión de los pacientes:Leiro-Fernández, Núñez-Delgado, Botana-Rial, Fernández-Villar.
Realización USEB-PTB: Leiro-Fernández, Fernández-Villar, Núñez-Delgado, Botana-Rial.
Análisis de Anatomía Patológica: Tardío-Baiges, González-Piñeiro.
Realización de las determinaciones moleculares: De Chiara, Valverde-Pérez.
Análisis e interpretación de los datos: Leiro-Fernández, De Chiara, Fernández-Villar, Leiro-Fernández, González-Piñeiro, Tardío-Baiges.
Análisis estadístico: De Chiara, Leiro-Fernández, Fernández-Villar.
Elaboración del manuscrito: De Chiara, Leiro-Fernández, Fernández-Villar.
Revisión crítica del manuscrito: Valverde-Pérez, Núñez-Delgado, Botana-Rial.
Supervisión del estudio: Fernández-Villar.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.







