


El síndrome de Kartagener es una enfermedad genética poco frecuente que se hereda de forma autosómica recesiva, con una afectación progresiva del sistema respiratorio y situs inversus. Aunque el tratamiento de los pacientes con el síndrome sigue siendo poco claro y las pruebas disponibles son limitadas, es importante su seguimiento con una asistencia adecuada y compartida. En el presente informe se describe un caso clínico del síndrome en una mujer de 25 años de edad. La tomografía computarizada demostró dextrocardia y bronquiectasias. La radiografía simple y la ecografía abdominal confirmaron un situs inversus total. Después de 7 años, se obtuvieron resultados satisfactorios del tratamiento: la función pulmonar mejoró y en la exploración radiológica no se demostraron cambios. En el presente artículo se describe la compleja interrelación entre la variación genética y un tratamiento inespecífico apropiado del síndrome.
Kartagener's syndrome is a rare autosomal-recessive genetic disease with progressive damage of the respiratory system and situs inversus. Although the management of patients with Kartagener's syndrome remains uncertain and evidence is limited, it is important to follow up these patients with an adequate and shared care system. This report presents a clinical case of Kartagener's syndrome in a 25-year-old woman. Computed tomography showed dextrocardia and bronchiectasis. Abdominal X-ray and ultrasound confirmed situs inversus totalis. After 7 years, good treatment results were achieved: lung function improved and radiological findings showed no changes. The present case discusses the complex interrelationship between the genetic variation and a proper nonspecific management of Kartagener's syndrome.
El síndrome de Kartagener es una enfermedad autosómica recesiva que se manifiesta principalmente por una afectación del movimiento ciliar1. El síndrome forma parte de un grupo mayor de enfermedades a las que se hace referencia como discinesias ciliares primarias (DCP). Aunque la enfermedad se hereda en un patrón autosómico recesivo, y se han reconocido algunos defectos genéticos específicos, está claro que el síndrome manifiesta una heterogeneidad genética sustancial2. La incidencia del proceso es de 1-2/30.000 nacimientos3. Manes Kartagener, neumólogo que trabajaba en Zurich, describió por primera vez la tríada de sinusitis, bronquiectasias y situs inversus en 19334. El apellido del Dr. Kartagener es de origen sefardí (judío español) y deriva de la ciudad española de Cartagena, que, a su vez, proviene de la ciudad fenicia de Cartago4.
Los síntomas son consecuencia de la motilidad defectuosa de los cilios de las vías respiratorias2,3. Las infecciones pulmonares recurrentes se deben a la afectación del transporte mucociliar en las vías respiratorias, lo que da lugar a la estasis de moco en los bronquios1,5. Hasta el momento del diagnóstico, acontece una lesión pulmonar progresiva y sustancial3,5. En niños más mayores y adultos con discinesia ciliar primaria se han descrito 3 enfermedades de las vías respiratorias inferiores: neumonía, bronquiectasias y asma6. Aunque el tratamiento de pacientes con el síndrome sigue por dilucidar, es importante controlar las infecciones pulmonares crónicas y el deterioro de la función pulmonar. Los esfuerzos para identificar las interacciones entre los factores genéticos y los determinantes ambientales podrían traducirse en mayores conocimientos sobre la patogenia de las bronquiectasias.
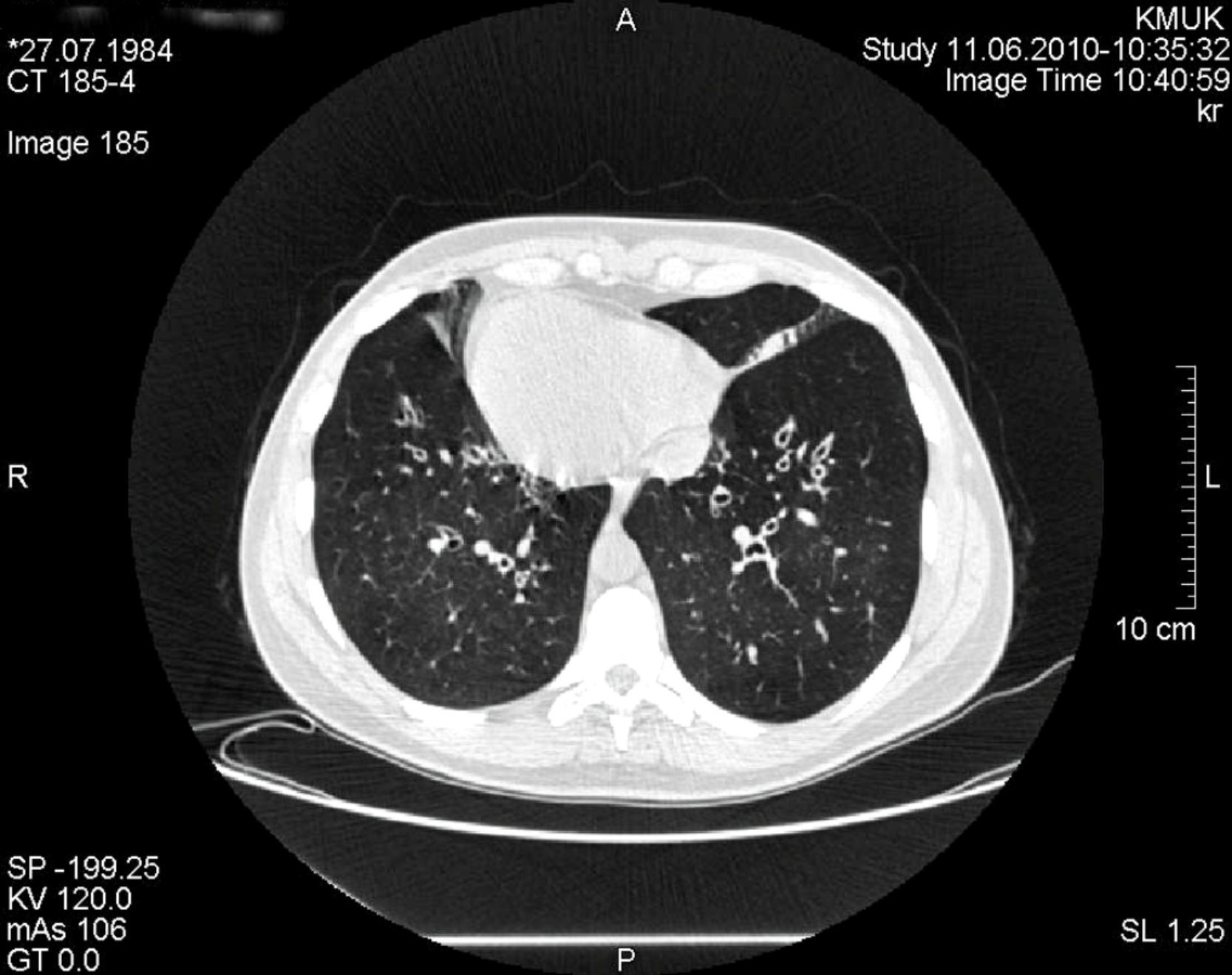
Caso clínicoSe presenta un caso clínico de síndrome de Kartagener en una mujer de 25 años de edad. La paciente refería tos y expectoración. Un examen del sistema respiratorio reveló crepitantes gruesos con roncus dispersos por el tórax. El latido de la punta se localizaba a la derecha, en posición lateral derecha. El hígado era palpable en el hipocondrio izquierdo. La tomografía computarizada (TC) de tórax demostró dextrocardia y bronquiectasias bilaterales (fig. 1). Las radiografías de senos demostraron inflamación de la mucosa nasal e hipertrofia mucosa de los senos maxilares, indicativo de rinosinusitis crónica. El aclaramiento mucociliar se valoró con la prueba de la sacarina, que deparó un tiempo de 48min (normal, <15min). La espirometría demostró una obstrucción leve: volumen espiratorio máximo en el primer segundo (VEMS), 93% del valor de referencia (3,27l); capacidad vital forzada (CVF), 110% (4,40l); VEMS/CV, 74%. Analizamos otros marcadores genéticos, que son importantes para la protección pulmonar: alfa-1 antitripsina (AAT), una antiproteasa, y la inmunoglobulina A (IgA) secretora. El genotipo AAT de la paciente era de tipo salvaje (PiMM) y la concentración de AAT se encontró en el límite superior del intervalo normal, en 2g/l. El valor de IgA inmunoprotectora también era alto, de 3,5g/l.

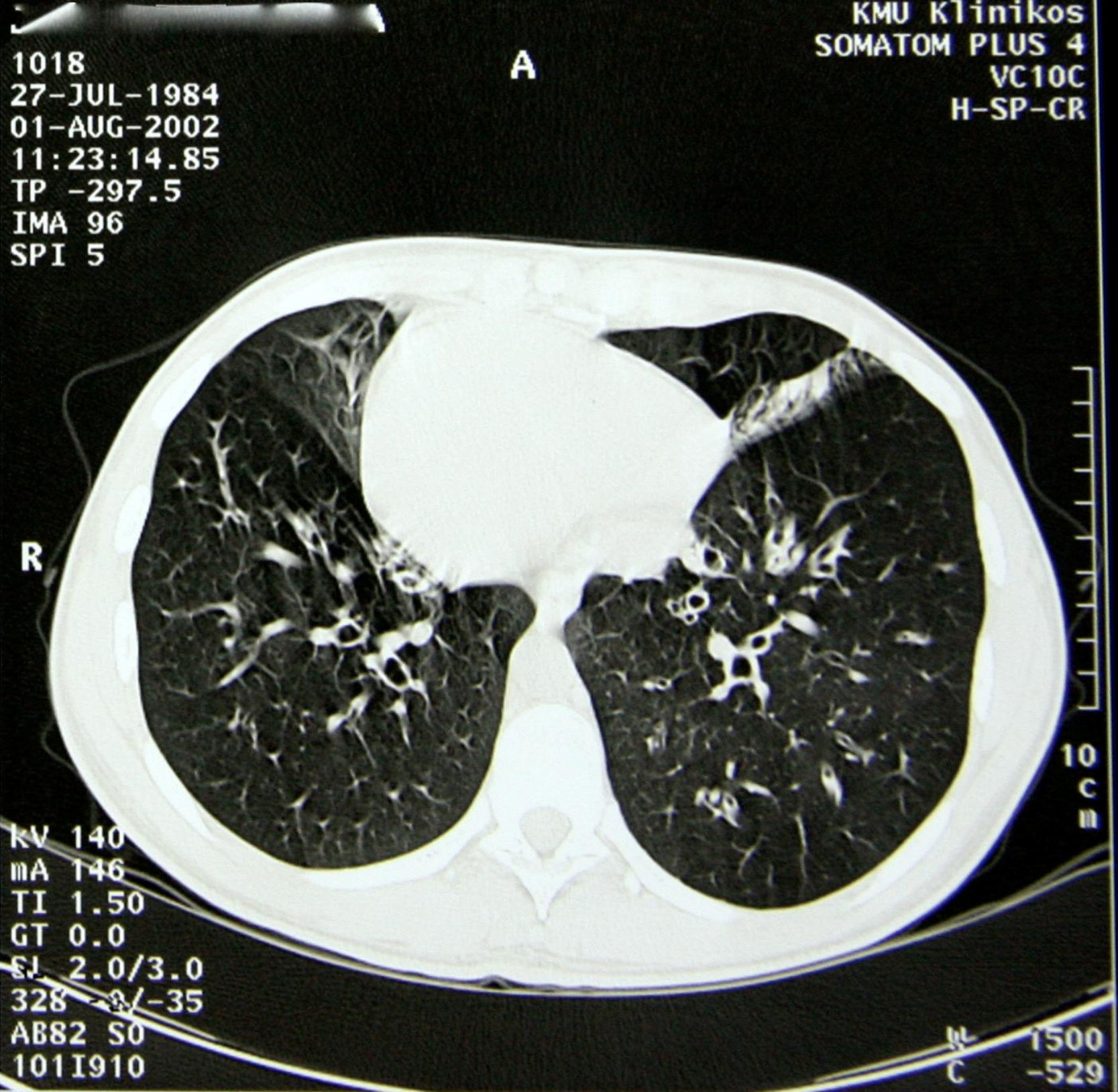
Entre los antecedentes patológicos de la paciente destacaban infecciones pulmonares de repetición, sinusitis crónica y otitis crónica desde la primera infancia. A los 14 años de edad se estableció el diagnóstico de síndrome de Kartagener. El diagnóstico se basó en el cuadro clínico y radiológico. El examen de cribado de la genealogía de la familia de la paciente no identificó otros casos del síndrome. En el momento del diagnóstico la espirometría demostró obstrucción bronquial de los bronquios de primer orden: VEMS, 88% del valor de referencia (3,08l); CVF, 111% (4,45l); VEMS/CV, 69%. Empezando a los 18 años de edad, la paciente recibió un seguimiento de neumólogos y genetistas. La TC del tórax (fig. 2) reveló dextrocardia, fibrosis pulmonar y múltiples bronquiectasias en lóbulos superiores. La radiografía simple y la ecografía abdominal confirmaron un situs inversus total (hígado izquierdo, burbuja gástrica y bazo a la derecha). En el examen microbiológico, los patógenos más habituales aislados fueron Haemophilus influenzae, Pseudomonas aeruginosa y Serratia marcescens.

Para el tratamiento de la obstrucción bronquial se administraron broncodilatadores inhalados de acción prolongada (agonistas β2 [salbutamol] a una dosis de 50μg/día). El tratamiento de las exacerbaciones persistentes de las bronquiectasias consistió en tandas breves de 10-14 días de antibióticos y mucolíticos (la bromhexina oral, a dosis de 16mg 3 veces al día, fue eficaz). La tasa media anual de infecciones de las vías respiratorias inferiores era de 2,5. Se aplicaron a diario fisioterapia junto con ejercicio físico, se optimizó la nutrición y la paciente evitó los contaminantes ambientales (incluido el humo del tabaco). Todos estos métodos redujeron los síntomas y niveles de marcadores inflamatorios y mejoraron la calidad de vida de la paciente. La espirometría se repitió una vez al año. La función pulmonar no se deterioró durante los 7 años de seguimiento: para supervisar la progresión de la enfermedad, cada 2 años se efectuó una TC de tórax. Los hallazgos radiológicos no demostraron cambios y la paciente no había desarrollado nuevas bronquiectasias (fig. 1).
DiscusiónLas características clínicas del síndrome de Kartagener son tos productiva, infecciones de las vías respiratorias, sinusitis, otitis media e infertilidad7,8. En las DCP el fenotipo clínico es amplio y se superpone con otras enfermedades crónicas de las vías respiratorias8. El defecto es congénito y los síntomas se presentan desde los primeros años de vida, lo que destaca la importancia de que los pediatras conozcan esta enfermedad como diagnóstico diferencial sustancial aunque poco frecuente en niños con síntomas recurrentes de las vías respiratorias superiores e inferiores5.
En la paciente descrita se estableció el diagnóstico de síndrome de Kartagener a los 14 años de edad. Con frecuencia, el diagnóstico de DCP se retrasa hasta la segunda infancia o la edad adulta como consecuencia de la naturaleza heterogénea de la enfermedad, la falta de conocimientos de los médicos sobre las características de la enfermedad y la experiencia técnica necesaria para un diagnóstico preciso3,8. Además, el diagnóstico de DCP puede retrasarse, ya que el síndrome, caracterizado por bronquitis, sinusitis y otitis, se confunde fácilmente con las infecciones comunes. El retraso en el reconocimiento de la enfermedad puede traducirse en consecuencias adversas para el paciente, desde un punto de vista de un programa insuficiente de asistencia o un tratamiento inapropiado8,9.
Se ha encontrado que otros marcadores genéticos detectados en los pacientes (AAT e IgA) son protectores (debido a la concentración elevada). La función primaria de AAT es inhibir la elastasa neutrófila en el intersticio pulmonar y el espacio alveolar. El aumento de los valores séricos de AAT puede reflejar un cambio beneficioso del equilibrio proteasas-antiproteasas, el elemento fundamental de la vía fisiopatológica que media el efecto del déficit congénito de AAT en la enfermedad pulmonar obstructiva crónica (EPOC) y las bronquiectasias. La detección de valores más altos de AAT y de IgA fue independiente del estado inflamatorio (los valores de proteína C reactiva eran normales). Formulamos la hipótesis de que estos factores protegerían frente a la infección y a la progresión adicional de las bronquiectasias.
El tratamiento respiratorio de las bronquiectasias consiste en una supervisión respiratoria a intervalos regulares, el aclaramiento de las vías respiratorias con combinaciones de fisioterapia y ejercicio físico, y un tratamiento agresivo de las infecciones de las vías respiratorias superiores e inferiores10. La paciente descrita recibió un amplio espectro de métodos curativos: tandas breves de antibióticos, mucolíticos, broncodilatadores de acción prolongada y fisioterapia diaria. En general, los antibióticos se usan de forma aguda con la exacerbación de la enfermedad y se prescriben de acuerdo con el crecimiento bacteriano en el último cultivo de esputo. El objetivo del tratamiento debe ser la prevención de una lesión pulmonar crónica y de las bronquiectasias11. Los pilares dobles del tratamiento respiratorio son la antibioterapia y la fisioterapia torácica. La fisioterapia es esencial para mejorar el aclaramiento de las vías respiratorias con el objetivo de retrasar el inicio y la progresión de la enfermedad obstructiva de las vías respiratorias4,10. El ejercicio físico puede contribuir al aclaramiento del esputo. Se ha demostrado que el ejercicio físico es un mejor broncodilatador que el uso de broncodilatadores en las DCP9. En general, se considera que el pronóstico es favorable, habitualmente con una esperanza de vida normal. Una importante parte de las visitas clínicas a intervalos regulares debe ser la monitorización de la progresión de la enfermedad pulmonar3. La paciente descrita fue seguida con visitas a intervalos regulares cada 6 meses, incluidas visitas adicionales durante las exacerbaciones.
El presente caso clínico demostró un curso clínico no progresivo de las bronquiectasias incluso en el caso de un síndrome de Kartagener congénito, lo que, por lo tanto, indica que la progresión de las bronquiectasias es una interrelación compleja entre la variación genética y un tratamiento inespecífico apropiado.







