


Introducción
Al menos el 90% de los pacientes diagnosticados de enfermedad pulmonar obstructiva crónica (EPOC) han sido fumadores crónicos1. La inclusión de la respuesta inflamatoria frente a las partículas y los gases tóxicos en la propia definición de la EPOC consensuada por la Global Initiative for Obstructive Lung Disease (GOLD)2 refleja la importancia central de la inflamación en los mecanismos patogénicos de la enfermedad. En esta respuesta inflamatoria, las acciones de las células efectoras y sus mediadores moleculares se imbrican de forma compleja con otros factores, tales como el estrés oxidativo, resultante de la exposición al humo del tabaco y de la propia inflamación, la acción de las proteasas sobre la matriz extracelular y las alteraciones de la capacidad de respuesta reguladora frente a todos estos agentes. Sin embargo, debe tenerse presente que sólo el 15-20% de los fumadores desarrolla EPOC1. Los mecanismos biológicos que determinan las diferencias entre los fumadores que desarrollan EPOC y aquellos que no lo hacen han sido un asunto central en este campo, y son un interrogante que continúa abierto. La búsqueda de determinantes genéticos de predisposición o protección frente a la enfermedad ha sido y sigue siendo un frente obvio de investigación, que ha resultado en la identificación hasta la fecha de numerosos polimorfismos alélicos de genes diversos, lo que confiere al desarrollo de la EPOC un trasfondo poligénico complejo3. Al menos algunas de estas variaciones alélicas hacen que los genes correspondientes actúen como factores modificadores de respuesta frente a la inflamación, el estrés oxidativo y la actividad proteolítica tisular. Como vía final común, pero altamente variable en sus formas fenotípicas, se producen alteraciones estructurales ("remodelación") que prestan sustrato fisiopatológico al deterioro de la función pulmonar que ocurre en los sujetos que desarrollan EPOC.
Bases inmunológicas de la EPOC. Inflamación
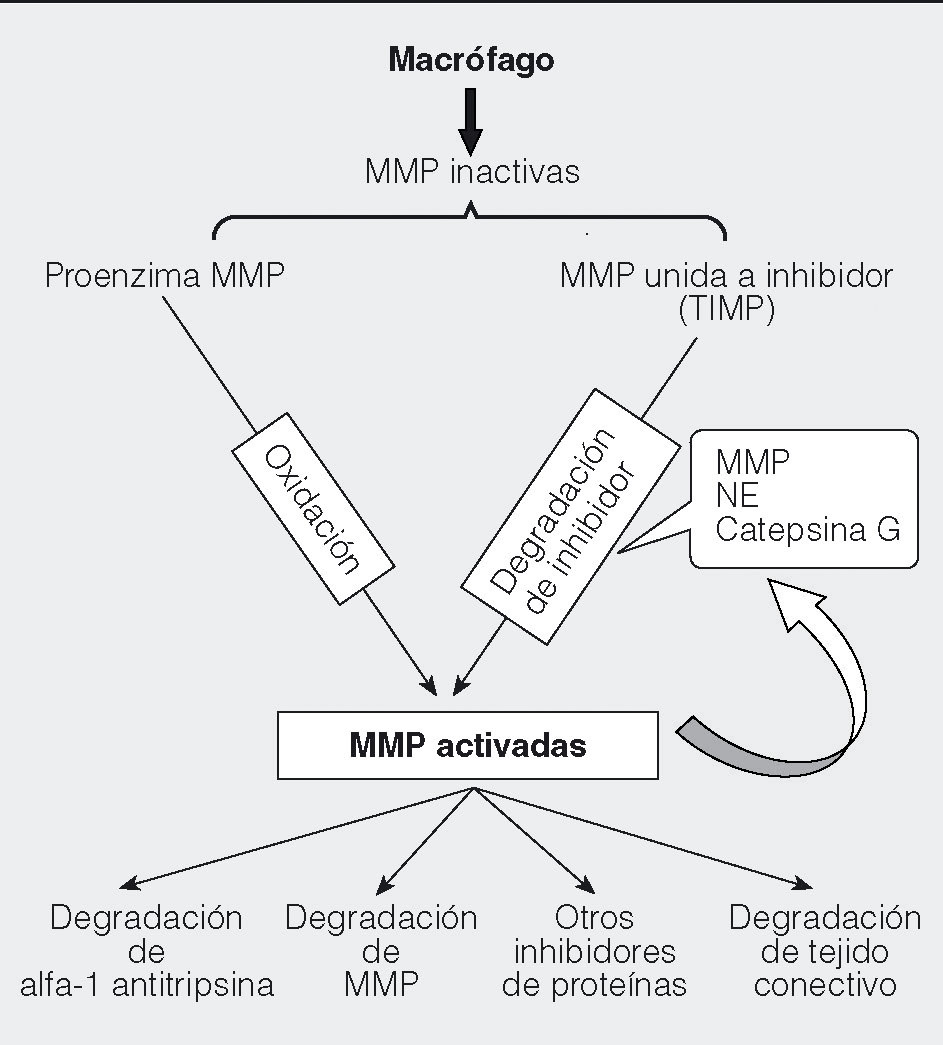
Una respuesta temprana frente a las toxinas inhaladas es el reclutamiento de macrófagos (fig. 1) a los pulmones, donde realizan la fagocitosis y la destrucción de noxas diversas, y seguidamente pueden ser eliminados por el arrastre ciliar del epitelio respiratorio. En los fumadores, se ha observado un incremento de la población de macrófagos en las vías respiratorias como respuesta a las toxinas del humo del tabaco4. En pacientes con EPOC, el número de macrófagos en lavado broncoalveolar (BAL) se encuentra elevado, como consecuencia del alto reclutamiento de estas células hacia las vías respiratorias, así como de un sistema de limpieza de macrófagos defectuoso, debido a la disfunción ciliar que el tabaquismo produce. Esta acumulación de macrófagos se correlaciona con la gravedad y la progresión de la enfermedad5, lo que indica un papel importante de estas células en la patogenia de la EPOC. Adicionalmente a sus funciones de eliminación de productos tóxicos y secreción de diversos metabolitos y mediadores, su capacidad de presentación antigénica podría desempeñar algún papel en la respuesta inmunitaria asociada a la EPOC. Entre los productos liberados por los macrófagos se encuentran especies de oxígeno reactivo (ROS), factores quimiotácticos, citocinas inflamatorias, constrictores del músculo liso, activadores de las glándulas mucosas y metaloproteasas (MMP). En combinación, todos estos componentes pueden dar lugar a una degradación de proteínas similar a la que llevan a cabo las enzimas de los neutrófilos6, y facilitar la migración de otros leucocitos a los tejidos dañados. La síntesis y la secreción de MMP por los macrófagos está regulada en respuesta al medio ambiente, según la presencia de citocinas, endotoxina, factores de crecimiento y actividad fagocítica (fig. 2). Se liberan en forma inactiva o unidas a inhibidores, y necesitan activarse mediante proteólisis del inhibidor u oxidación de la enzima latente7. Una vez activadas, pueden regular la actividad de las citocinas mediante su degradación o regular otras proteinasas como la alfa-1 antitripsina y generar una cascada de activación. En los macrófagos de pacientes con EPOC hay elevados valores de MMP1, 2, 9 y 148-11.

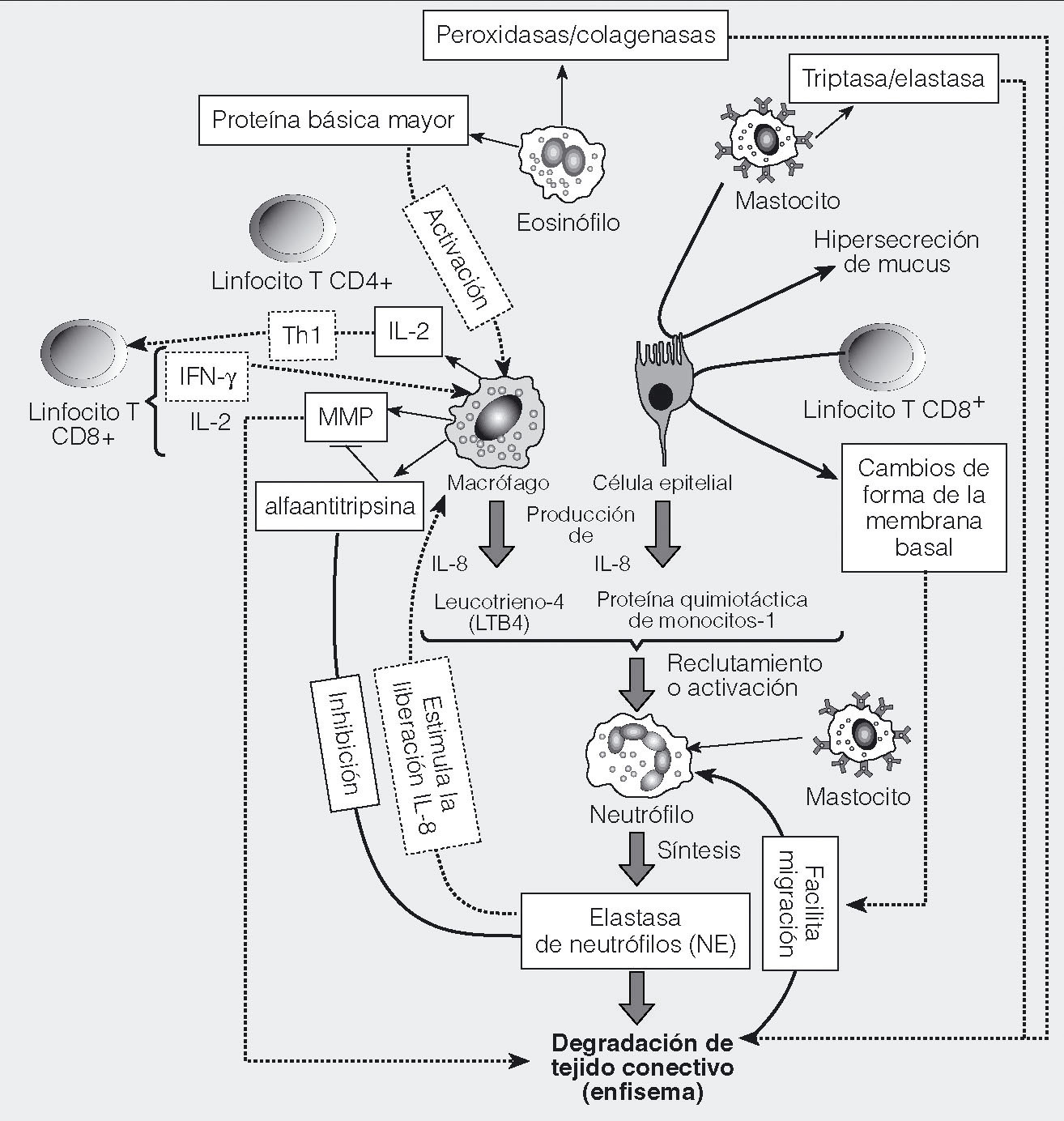
Fig. 1. Células inmunitarias participantes en la enfermedad pulmonar obstructiva crónica (EPOC). Las células de primera línea de defensa frente a los agentes tóxicos contenidos en el humo del tabaco son los macrófagos y neutrófilos, y las propias células epiteliales. Está menos claro el papel de los eosinófilos y los mastocitos, aunque se ha observado que los eosinófilos están aumentados en las exacerbaciones. Los macrófagos y los neutrófilos son la fuente principal de secreción de diversas proteasas que, en conjunto, tienen capacidad para degradar la matriz extracelular. Adicionalmente, la actividad antiproteasa puede estar disminuida como consecuencia del estrés oxidativo, y por diversas formas de déficit genético en ciertos individuos. El desequilibrio proteasa/antiproteasa es la teoría más tradicional sobre la patogenia del enfisema, y la evidencia actualmente acumulada indica que es un mecanismo contribuyente. Macrófagos y neutrófilos segregan, asimismo, quimiocinas y citocinas, que inducen el reclutamiento de más células y amplifican la inflamación en la EPOC. Datos recientes indican que el sistema inmunitario adaptativo puede estar implicado en una respuesta autoinmunitaria, que origina parte de la infiltración inflamatoria e induce apoptosis de células estructurales. Los efectores de esta respuesta serían linfocitos T CD8+ citotóxicos y linfocitos T CD4+, con activación tipo Th1, una de cuyas funciones es estimular a las células T CD8+. El origen de la activación linfocitaria es desconocido, pero pueden participar en ella la liberación de antígenos intracelulares por daño celular, la generación de autoantígenos por modificaciones proteínicas inducidas por reactantes químicos del humo del tabaco, la infección adenoviral latente, la presentación antigénica por células epiteliales y otros tipos celulares, y la secreción de citocinas tipo Th1 como la interleucina (IL) 12 y el interferón (IFN) Υ.

Fig. 2. Modo de actuación de las metaloproteasas (MMP). Las MMP se liberan inactivas en forma de proenzima, o de enzima unida a inhibidores tisulares (TIMP). Su activación se produce mediante la oxidación de la proenzima o mediante la degradación de los inhibidores. Una vez activadas, su función proteasa degrada a otras proteínas tanto estructurales (matriz extracelular) como reguladoras (alfa-1 antitripsina, otras MMP y otros inhibidores de proteínas).
Los neutrófilos, otro tipo celular de la primera línea de defensa inmunitaria inespecífica, también se presentan en número aumentado en la EPOC12-14, y particularmente elevados durante las exacerbaciones graves15. El aumento de neutrófilos, en general, se ha detectado mediante procedimientos que muestrean el lumen de las vías respiratorias (BAL y esputo), y sin embargo, es escaso en la infiltración tisular en la pared. Una posible explicación para esta discrepancia podría ser una rápida migración de los neutrófilos a través de los tejidos y su acumulación en el lumen16. Los neutrófilos son fuente de diversos mediadores, incluyendo ROS, citocinas inflamatorias, mediadores lipídicos, péptidos antibacterianos, proteasas y MMP, como la colagenasa de neutrófilos (MMP8) y la gelatinasa B (MMP9), que tienen capacidad para la degradación de gran parte de los componentes de la matriz extracelular17. Secretan, además, tipos propios de proteinasas neutrofílicas con gran capacidad de degradación de la elastina y que son inhibidas por la alfa-1 antitripsina. Estos datos, junto con la observación de que la frecuencia de déficit parcial o grave de alfa-1 antitripsina en sujetos con EPOC es superior a la de la población general18, indican un papel de los neutrófilos en la aceleración que el tabaquismo produce en el desarrollo de enfisema asociado al déficit de esta antiproteasa. Sin embargo, la contribución de los neutrófilos a los mecanismos de destrucción tisular es discutida, dado que se ha observado una correlación inversa entre la cantidad de neutrófilos y el desarrollo de enfisema5.
La relevancia de los eosinófilos en la EPOC es discutida, debido a la discordancia entre los datos que indican o no su aumento19,20, o que están presentes en forma inactiva14. Es destacable que el número de eosinófilos se puede ver incrementado en la pared de las vías respiratorias durante las exacerbaciones de la EPOC en sujetos no atópicos, junto con el aumento de la expresión de las citocinas interleucina (IL) 4 y 5 y de las quimiocinas atractoras de eosinófilos RANTES (regulated on activation, normal T-cell expressed and secreted), la eotaxina y la MCP (monocyte chemoattractant protein) 421. La presencia de eosinófilos se presenta como un predictor de respuesta al tratamiento esteroideo22,23. En la EPOC se ha descrito también un aumento de mastocitos24, que podrían estar implicados en el reclutamiento de neutrófilos mediante la liberación de factores quimiotácticos, el daño tisular por la acción de diversas enzimas (triptasa, quimasa y elastasa) y la hipersecreción de moco por la acción de la quimasa25.
Durante décadas, el conocimiento ganado sobre la biología de los macrófagos y los neutrófilos, y su capacidad para segregar varias enzimas con potencial para digerir componentes intersticiales del parénquima pulmonar, ha fortalecido una teoría clásica sobre la patogenia de la EPOC nacida del hallazgo de la asociación entre el déficit genético de alfa-1 antitripsina y el desarrollo de enfisema pulmonar26,27. Se trata de la teoría del desequilibrio "proteasa-antiproteasa", según la cual el enfisema pulmonar es resultado de la digestión elastolítica de la matriz extracelular debido a las proteasas, cuya acción puede no ser debidamente contrarrestada, ya sea por déficit genético de antiproteasas, por liberación excesiva de proteasas por células inflamatorias, o bien por una combinación de ambos. De este modo, macrófagos y neutrófilos se han situado tradicionalmente en el centro de los mecanismos patogénicos de la EPOC en fumadores. Sin embargo, investigaciones desde un nuevo ángulo, en la década de los años noventa, originaron un importante giro de conceptos sobre el modo en que el sistema inmunitario, y más específicamente su vertiente adaptativa, está implicado en los mecanismos de la EPOC. La observación de que los fumadores y los fumadores con enfisema tienen infiltración inflamatoria por células T, cuyo número se correlaciona con el grado de destrucción del parénquima pulmonar5, condujo a generar nuevas hipótesis, al requerir una explicación para el papel de las células T en la EPOC. Este incremento de células T resultó deberse predominantemente a la subpoblación CD8+, tanto en las vías respiratorias centrales28 y periféricas19,29 como en el parénquima20. Estas observaciones, junto con el conocido papel de las células T CD8+ en la inducción de citólisis o apoptosis de células infectadas por virus30,31, condujo a plantearse un posible papel de la función citotóxica de las células T CD8+ en la patogenia de la EPOC, y del enfisema en particular. Estudios más recientes mostraron que las células T CD8+ se encuentran activadas en la EPOC, con expresión del receptor de quimioquinas CXCR3 y producción de interferón (IFN) Υ32, y secreción aumentada de perforinas y granzimas33,34. En consecuencia, la siguiente cuestión ha interrogado el origen de una respuesta inmunitaria adaptativa dirigida por antígenos en la EPOC, que resulta en la activación de células T. Una teoría propuesta es la existencia de infección adenoviral latente en los sujetos que desarrollan enfisema, con expresión de antígenos adenovirales en los neumocitos35. Otra hipótesis es la inducción de una reacción autoinmunitaria en los fumadores que desarrollan EPOC, frente a proteínas liberadas por la destrucción de la matriz extracelular y daño celular directo, más neoantígenos o proteínas modificadas por acción química de radicales libres, como resultado de los efectos tóxicos de la exposición al humo del tabaco36-38. La capacidad de las células T CD8+ antígeno-específicas para desencadenar inflamación autoinmunitaria frente al antígeno diana presentado por neumocitos se había demostrado experimentalmente, aunque en este modelo los ratones desarrollaron neumonitis intersticial con un defecto mecánico restrictivo39. En este trabajo no se investigó la inducción de apoptosis in vivo de neumocitos por las células T CD8+, y el diseño del modelo probablemente no habría facilitado el desarrollo de enfisema. En todo caso, y aun faltando hasta la fecha evidencia experimental, la idea sobre la implicación del sistema inmunitario adaptativo en una respuesta autoinmunitaria en la EPOC se ha visto reforzada por el reciente hallazgo40 de un aumento en las biopsias bronquiales y el lavado broncoalveolar de sujetos con EPOC, de células T CD4+ con activación de la vía de señalización mediada por STAT-4 (signal transducer and activator of transcription 4) y secreción de IFN-g. Estos datos indican la existencia en el EPOC de una respuesta inmunitaria adaptativa con activación de células T CD4+ tipo helper-1 (Th1), de las cuales una conocida función es el soporte de las respuestas citotóxicas de células T CD8+. Respecto de las células T natural killer (NK), una subpoblación citotóxica especializada en eliminar células transformadas o infectadas por virus, los datos son controvertidos acerca de su presencia aumentada41 o no42 en la EPOC.
Estrés oxidativo
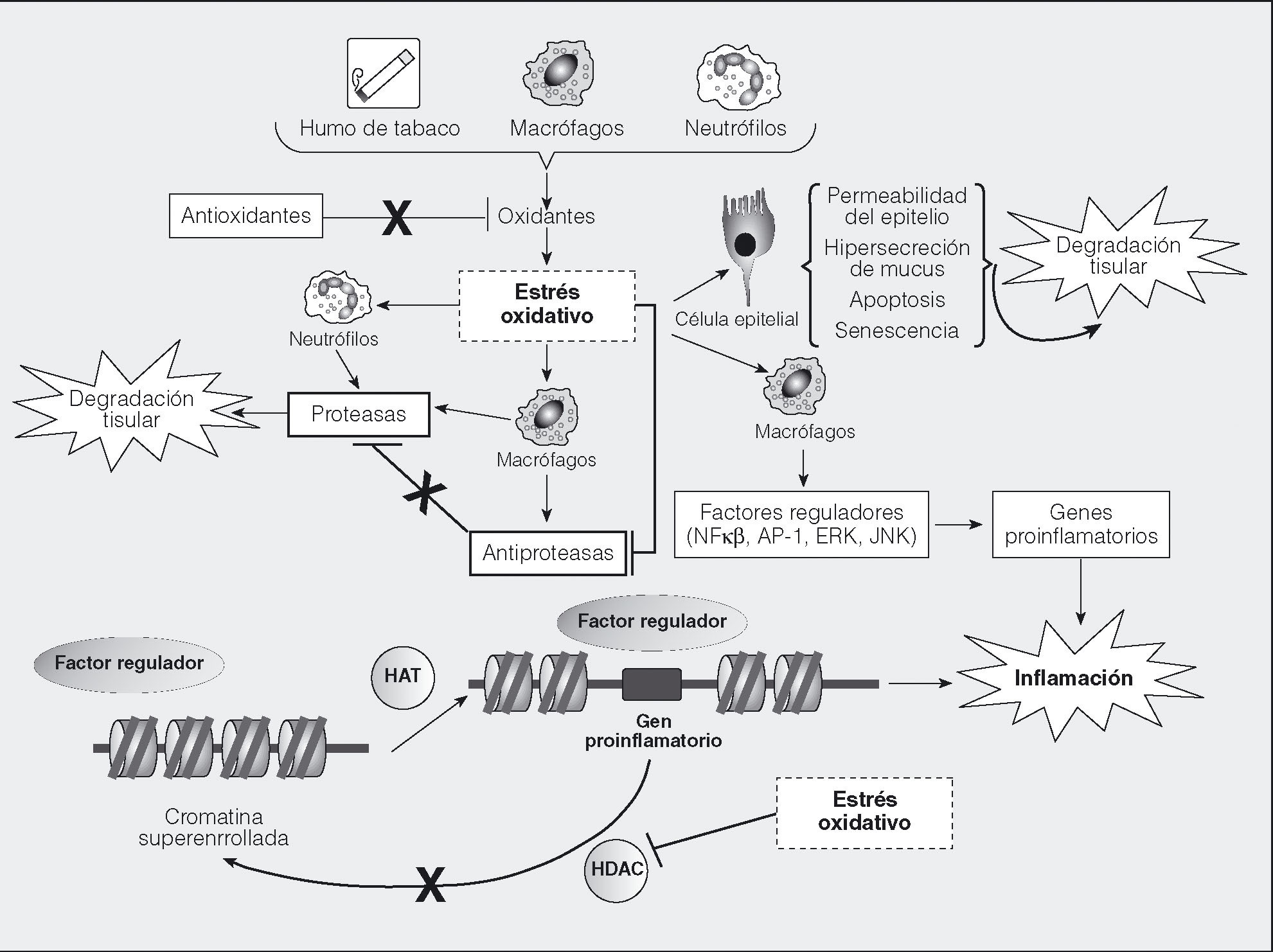
Los radicales libres u oxidantes son producidos por la actividad de una gran variedad de oxidasas durante muchos procesos celulares normales y están caracterizados por tener uno o más electrones no pareados, que hacen a estas moléculas altamente reactivas. Cuando un radical libre reacciona con una estructura molecular estable puede originar una reacción en cadena que generará nuevos radicales libres. La mayoría de las moléculas de los organismos vivos son susceptibles de reacción con los abundantes radicales libres presentes en la polución o en el humo de un cigarrillo. Los radicales libres se generan también en las reacciones inflamatorias y en el metabolismo celular, y tienen funciones fisiológicas, como la defensa frente a patógenos mediante la exposición a ROS y especies de nitrógeno reactivo (RNS) de bacterias fagocitadas43, la participación en vías de señalización y la regulación de la activación de diversas proteasas, factores de crecimiento y citocinas44. Los pulmones contienen nuestra mayor superficie de exposición al medio exterior; una gran superficie, por tanto, de interacción con los oxidantes contenidos en la polución y en el humo del tabaco, y con oxidantes de producción endógena por células inflamatorias (fig. 3). Frente a este reto oxidativo, los pulmones se protegen mediante sistemas antioxidantes. Cuando el balance oxidantes/antioxidantes se inclina hacia los oxidantes, se produce "estrés oxidativo", que genera daños mediante mecanismos que incluyen modificaciones de aminoácidos y proteínas, alteraciones en el ADN y activación de cascadas proinfamatorias, más el efecto amplificador del daño por los radicales oxidantes producidos por las propias células inflamatorias.

Fig. 3. Estrés oxidativo y sus efectos. Los radicales libres oxidantes están presentes en el humo del tabaco y también son producidos por las células de la respuesta inmunitaria. Si su carga desborda los sistemas de destoxificación antioxidante, se produce estrés oxidativo, que produce daño directo en las células estructurales, altera la producción de citocinas y proteasas por neutrófilos y macrófagos, e inhibe los sistemas antiproteasa. También afecta a la acción de las deacetilasas (HDAC) y acetilasas (HAT) de las histonas, parte de la maquinaria de remodelación de la cromatina, bloqueando las HDAC y favoreciendo la acetilación y la configuración desarrollada de la cromatina. Esta configuración facilita el acoplamiento al ADN de factores reguladores de la maquinaria transcripcional e induce la expresión de genes proinflamatorios. Asimismo, bloquea la acción terapéutica de los corticoides, uno de cuyos mecanismos es el reclutamiento de HDAC al núcleo celular.
Existe abundante evidencia acumulada sobre la existencia de estrés oxidativo en los fumadores y en sujetos con EPOC en el pulmón y a escala sistémica45-47, así como datos que relacionan el incremento de marcadores indirectos de estrés oxidativo con el desarrollo de limitación obstructiva al flujo aéreo48,49. El humo del cigarrillo induce expresión diferencial de diversos genes implicados en la respuesta al estrés oxidativo en el epitelio bronquial, y estas diferencias resultan amplificadas en los fumadores que desarrollan EPOC50. Los oxidantes del humo del cigarrillo pueden estar implicados en las manifestaciones sistémicas del EPOC, como la disfunción de la musculatura esquelética y respiratoria y la pérdida de peso. El estrés oxidativo en estos sujetos está causado por diversos radicales libres, incluyendo aniones superóxido (O2) liberados por los macrófagos alveolares o derivados de reacciones xantina/xantina oxidasa51, óxido nítrico (NO)52, monóxido de carbono (CO), etano53 y productos de peroxidación lipídica, entre los que se incluyen derivados de la peroxidación del ácido araquidónico (isoprostanos)54,55.
Entre las consecuencias más directas del estrés oxidativo en los pulmones se encuentran daños en los epitelios que recubren los espacios aéreos, en células de tejidos conectivos y en el endotelio vascular. Experimentalmente, se ha mostrado que el estrés oxidativo causa un aumento de permeabilidad epitelial asociado a depleción de glutatión, una de las principales moléculas reductoras (antioxidante)56, y discinesia ciliar del epitelio respiratorio57. Se ha observado, también, una correlación entre el aumento de los marcadores de estrés oxidativo y la apoptosis de células endoteliales58 y, tras una exposición prolongada al humo del tabaco, la producción de un fenotipo de senescencia en los fibroblastos, caracterizado por una pérdida de la capacidad de división celular y de resistencia a la apoptosis59. En consecuencia, los fibroblastos son incompetentes para reparar el daño tisular asociado al desarrollo de enfisema. En los pulmones de sujetos con EPOC existen, asimismo, poblaciones de neutrófilos retenidos como consecuencia de una capacidad de deformación limitada, que resulta de un aumento de polimerización de actina debida a estrés oxidativo, que revierte mediante el uso de antioxidantes60. Los neutrófilos retenidos son activados, liberan ROS y proteasas, y contribuyen a la destrucción de la pared alveolar. Los radicales oxidantes inducen, asimismo, la liberación de quimiocinas y citocinas proinflamatorias, como la IL-8 y el factor de necrosis tumoral α (TNF-α)61, y activan mediadores de señalización intracelular y factores de transcripción reguladores de la expresión de genes implicados en la inflamación, como el factor nuclear kβ (NF-kβ), la proteína activadora 1 (AP-1), las cinasas ERK (extracellularly regulated kinase) y JNK (N-terminal c-Jun kinase), y las rutas de la proteincinasa activada por p3862. Se ha demostrado también que la infección adenoviral latente potencia la respuesta inflamatoria al humo del tabaco mediante un efecto sinérgico en el reclutamiento de macrófagos y células T63. La proteína E1A derivada del adenovirus se une a factores de transcripción y favorece su unión a las regiones reguladoras de sus genes diana, y por esta vía puede contribuir a aumentar la inflamación y hacer que las células que la poseen sean más sensibles al estrés oxidativo63. Adicionalmente a la contribución del estrés oxidativo a la activación leucocitaria y el aumento de la carga de proteasas, los radicales oxidantes inactivan antiproteasas como la alfa-1 antitripsina, y potencian el desequilibrio proteasa/antiproteasa64, y podrían inactivar genes implicados en respuestas de destoxificación antioxidante. En este sentido, se ha mostrado que la inactivación del factor de transcripción Nrt2 (nuclear factor, erythroid-derived 2), implicado en la regulación de varios genes de respuesta citoprotectora antioxidante, anticipa y acelera el desarrollo de enfisema en respuesta al humo del tabaco65.
Entre las consecuencias del estrés oxidativo, han atraído especial atención en el campo del EPOC sus efectos sobre la remodelación de la cromatina, un importante mecanismo de regulación de la expresión genética. En cada célula, una porción del ADN genómico está compactada o "superenrrollada" en forma de heterocromatina, que constituye un entorno de silenciamiento genético debido a la inaccesibilidad de la maquinaria transcripcional (factores de transcripción, ARN polimerasa y otros complejos moleculares). El enrollamiento de ADN en forma de heterocromatina, o su desenrollamiento para permitir la transcripción de genes, están regulados por modificaciones químicas en las histonas, entre las que se encuentra la desacetilación/acetilación por enzimas respectivas, que inducen el silenciamiento o la liberación de la expresión genética, respectivamente66. El estrés oxidativo afecta estos procesos promoviendo, por una parte, el estado acetilado de las histonas, lo que induce transcripción de genes entre los que se encuentran genes relacionados con la inflamación e inhibiendo, por otra, la actividad de las deacetilasas67-69. La gravedad de la EPOC se correlaciona con el grado de disminución de la actividad de la deacetilasa de las histonas y con el incremento de acetilación en el promotor del gen de la IL-8, y la transcripción de este gen68. Los efectos del estrés oxidativo sobre el balance entre la desacetilación y la acetilación de las histonas, como mecanismo de alteración de la expresión genética, han adquirido especial interés por su propuesta participación en la resistencia al tratamiento esteroideo que caracteriza a la EPOC. Un mecanismo de acción de los corticoides que se puede ver afectado o bloqueado por el estrés oxidativo consiste en el reclutamiento de deacetilasas de histona al núcleo celular, lo que induce el silenciamiento de genes inflamatorios70.
Cambios estructurales pulmonares en la EPOC. Estudio de la remodelación
La acción de las células del sistema inmunitario activadas, los mediadores inflamatorios y el estrés oxidativo conduce a diversos cambios estructurales en los tejidos pulmonares, denominados, en conjunto, remodelación, que constituyen en gran medida el sustrato fisiopatológico de las manifestaciones clínicas de la EPOC. Estos cambios afectan, en mayor o menor medida, según un amplio espectro fenotípico de la enfermedad, a las vías respiratorias, el parénquima y la vasculatura pulmonar. Un breve sumario de la histología del pulmón normal71,72 es conveniente como paso previo a un mejor entendimiento de estos fenómenos de remodelación tisular patológica. La pared de las vías respiratorias de conducción consta de una túnica mucosa, constituida por un epitelio cilíndrico ciliado seudoes-tratificado, en el que se intercalan células caliciformes productoras de moco, células neuroendocrinas, células basales relativamente indiferenciadas y terminaciones nerviosas. En las vías respiratorias más distales, el epitelio bronquial se convierte progresivamente en una capa de epitelio cilíndrico monoestratificado, no existen glándulas, la frecuencia de células caliciformes disminuye y aparecen las células bronquiolares de Clara. Las células del epitelio se asientan sobre una membrana basal, bajo la que subyace una región de tejido conjuntivo laxo denominada lamina propria, formada por un escaso contenido celular a base de fibroblastos, fundamentalmente, y matriz extracelular, compuesta por fibras de colágeno, fibras elásticas y otros componentes. La la-mina propria es un espacio de distribución de microvasculatura de la circulación bronquial sistémica y fibras nerviosas, y un lugar preferente de acumulación leucocitaria y edema, en situaciones de inflamación de las vías respiratorias. Por fuera de la túnica mucosa, se encuentra la túnica muscular, formada por haces de células musculares lisas en disposición helicoidal cruzada con ángulo variable, que conforma un patrón de distribución "geodésico". En los bronquios propiamente dichos, el cartílago se sitúa inmediatamente por fuera del músculo liso y pierde progresivamente su organización anular a lo largo de las primeras generaciones bronquiales, hasta desaparecer. Las generaciones subsiguientes de las vías respiratorias, libres de cartílago, se denominan bronquiolos. Externamente al músculo liso se encuentra la túnica adventicia de la vía respiratoria. Éste es otro espacio de tejido conjuntivo laxo, en cuyo límite externo se anclan las paredes alveolares del parénquima pulmonar, y constituye una región estructural de especial importancia en el balance elastomérico entre la vía respiratoria, que tiende a su reducción radial, y la red del parénquima pulmonar, que en condiciones normales "tira" de la vía respiratoria y tiende a mantenerla distendida. Este equilibrio se denomina interdependencia mecánica entre la vía respiratoria y el parénquima. La túnica adventicia aloja los acinos de las glándulas mucosas (denominadas también submucosas por su localización), cuyos conductos excretores vierten el contenido secretado a la luz de la vía respiratoria. Esta envoltura es, asimismo, un espacio conectivo de distribución de vasos de la circulación bronquial y nervios, se fusiona con la túnica adventicia de arterias vecinas de la circulación pulmonar, y constituye un importante espacio de tráfico leucocitario en situaciones de inflamación.
La generación más distal de vías respiratorias de conducción son los bronquiolos terminales. Cada bronquiolo terminal origina una o más generaciones de bronquiolos respiratorios, así denominados por presentar en su pared algunas discontinuidades que se abren a alvéolos pulmonares, y constituyen de este modo una región de transición entre el árbol de vías respiratorias de conducción y la región de intercambio de gases o parénquima pulmonar. Los bronquiolos respiratorios se abren finalmente a conductos alveolares que se distribuyen hacia atrios y sacos alveloares, y alvéolos. El haz formado por un bronquiolo terminal, sus ramificaciones y la porción de parénquima que ventila se denomina acino respiratorio. Los acinos se organizan en conjuntos de 3 a 5, formando unidades estructurales de parénquima pulmonar tabicadas por tejido conectivo, que se denominan lobulillos. Esta terminología es importante para la descripción de las distintas formas de enfisema pulmonar. Finalmente, las paredes alveolares están revestidas en un 95% de su superficie por un epitelio plano (neumocitos tipo I) especializado en formar parte de la interfase hemato-aérea. En el epitelio alveolar se incluyen, asimismo, células cuboideas, los neumocitos tipo II, especializados en la secreción de surfactante pulmonar. Entre las láminas de epitelio y sus respectivas membranas basales contrapuestas, se sitúa un fino espacio de tejido conectivo con una malla reticular de fibras de colágeno y elásticas, en el que se distribuye la red capilar de la circulación pulmonar.
Prácticamente todas las estructuras aquí descritas se alteran en la EPOC, en mayor o menor medida según el fenotipo de la enfermedad. Aunque muchos de estos cambios estructurales resultan obvios en un examen cualitativo de preparaciones anatomopatológicas, han sido las técnicas de morfología cuantitativa las que han permitido un análisis sólido de estas alteraciones y su puesta en relación con otras variables, tales como la presencia de células inflamatorias. Una de las mediciones que ofrece la morfología cuantitativa, aplicada a la cuantificación del enfisema pulmonar, es la densidad volumétrica o "volumen en volumen". Se trata de determinar el volumen de tejido de parénquima pulmonar que existe por unidad cúbica de volumen total. Esta variable correspondiente a una estructura tridimensional se puede inferir a partir de mediciones bidimensionales sobre preparaciones microscópicas convencionales ("morfometría"), o se puede obtener a partir de mediciones volumétricas directas en preparaciones gruesas ("estereología"). El cuerpo matemático y la descripción de los procedimientos sobre los que se asientan estas técnicas son un tema que excede el propósito de esta revisión73-76. Respecto a las vías respiratorias, es frecuente la necesidad de inferir la cantidad de un componente tisular o un tipo celular determinados. En este caso, es necesario corregir dicha medición por el tamaño de la vía respiratoria. La referencia tomada con más frecuencia es el perímetro de la membrana basal en las vías respiratorias seccionadas transversalmente, que se considera la dimensión más constante en una vía respiratoria, con independencia de su estado de constricción o relajación77. Las preparaciones más apropiadas para estudios de morfología cuantitativa son aquéllas obtenidas de pulmones o lóbulos pulmonares completos, donde es posible muestrear regiones amplias de parénquima pulmonar y vías respiratorias completas, o sectores de arco de su pared íntegra. Los estudios post mórtem se han utilizado para esta finalidad, pero están limitados por factores técnicos relativos a la conservación de los tejidos, especialmente de estructuras antigénicas específicas, si se pretende realizar un análisis inmunohistoquímico, dado que numerosos determinantes antigénicos se degradan rápidamente. Por esta razón, las preparaciones óptimas son piezas de resección quirúrgica, tratadas con agentes fijadores de forma inmediata, cuando el tejido está aún vivo. La utilización de lóbulos o pulmones de resección quirúrgica facilita, además, un procedimiento de importancia crítica para la normalización adecuada de los datos cuantitativos, que es fijar el pulmón o el lóbulo reseccionado, inflado a presión estándar con el propio agente fijador (comúnmente formalina) insuflado por vía bronquial, utilizando una bomba de recirculación para el mantenimiento de una presión constante. Fijar los pulmones inflados a presión estandarizada es, asimismo, esencial en los modelos animales de enfermedad experimental. Otro procedimiento de muestreo in vivo es la biopsia bronquial. La biopsia ha aportado abundante información sobre fenómenos de remodelación en la zona epitelial y subepitelial (lamina propria), pero su versatilidad se ve limitada por su profundidad (sólo se logra un muestreo parcial de la túnica de músculo liso) y una mayor dificultad para la normalización de datos cuantitativos. Finalmente, mediante biopsia transbronquial, se han podido evaluar procesos de remodelación en los bronquiolos más periféricos.
Alteraciones estructurales de las vías respiratorias
En los fumadores, en asociación con la limitación al flujo aéreo, se ha descrito infiltración inflamatoria y cambios estructurales en las vías respiratorias, tanto centrales como periféricas (fig. 4). Las alteraciones más periféricas, en los bronquiolos, han atraído especial atención, ya que se ha demostrado que ésta es la principal localización de la resistencia al flujo aéreo78, y que los fumadores jóvenes, bastantes años antes del posible desarrollo de EPOC, presentan inflamación en esta área79. Las alteraciones descritas en los bronquiolos incluyen hiperplasia de células caliciformes (metaplasia mucosa), aumento de la masa de músculo liso y fibrosis80,81. La hiperplasia de células caliciformes se asocia a hipersecreción mucosa y oclusión luminal por tapones de moco. El músculo liso aumentado, la fibrosis y la propia presencia de inflamación contribuyen a la obstrucción al aumentar el grosor de la pared bronquiolar y, también, probablemente, al desacoplar la vía respiratoria de la tracción elástica del parénquima. Adicionalmente, se describió en fumadores la rotura de los anclajes alveolares asociada a la inflamación bronquiolar y a pérdida de retracción elástica, lo que indica que hay un mecanismo que contribuye a la obstrucción en estadios tempranos del desarrollo de EPOC82.

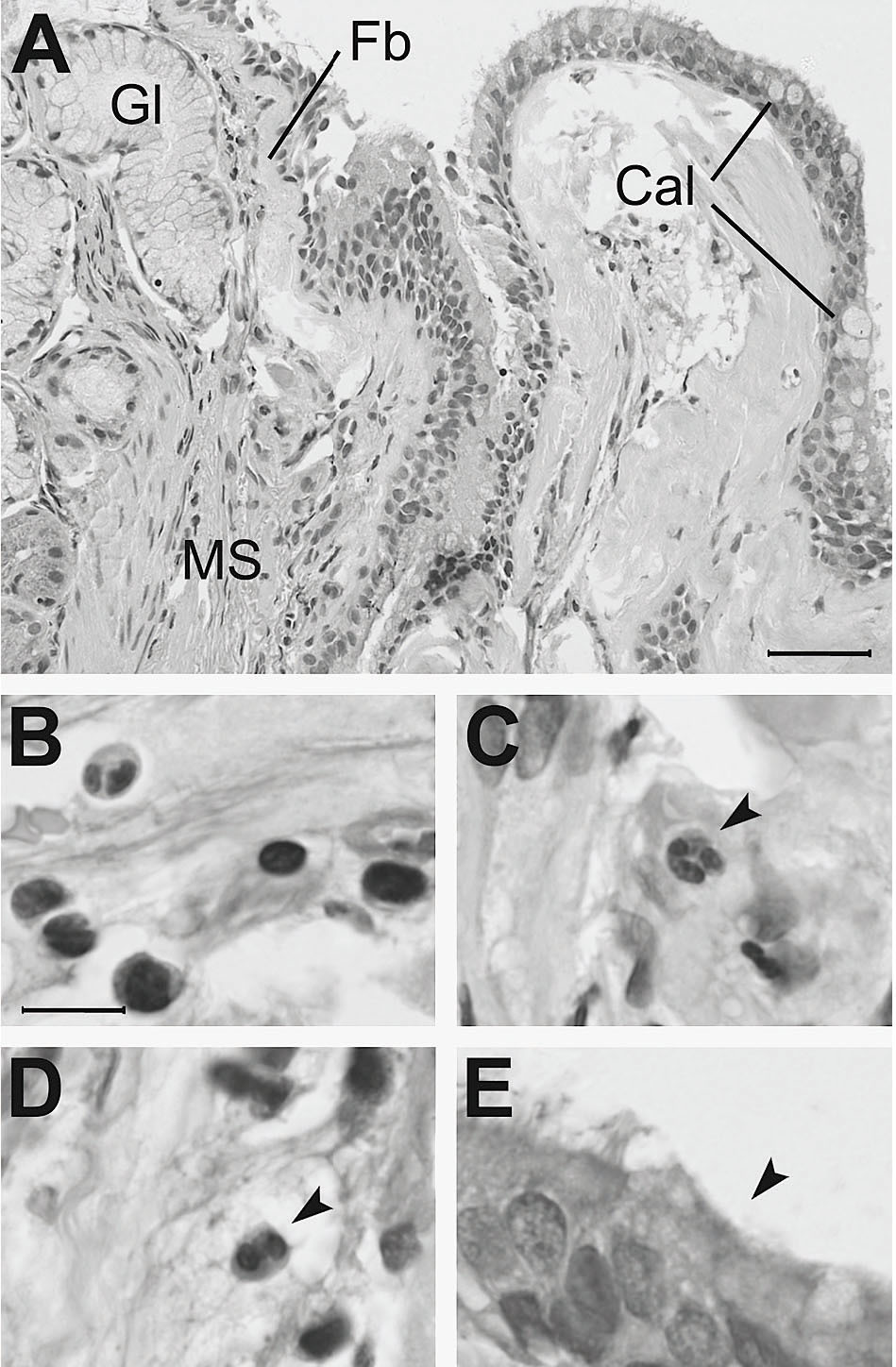
Fig. 4. Biopsia bronquial de paciente con enfermedad pulmonar obstructiva crónica. A) La sección histológica muestra: acinos glandulares mucosos (Gl) y músculo liso (MS) abundantes y muy cercanos al epitelio, un signo que refleja el crecimiento de estas estructuras; áreas de fibrosis de localización subepitelial (Fb), que se observan como depósitos hialinos prácticamente acelulares, y abundantes células caliciformes (Cal), con aspecto hipertrófico. Estos cambios estructurales se acompañan de inflamación con infiltración mononuclear (B), presencia de neutrófilos (C) y eosinófilos (D). Se observan también zonas de destrucción de los cilios del epitelio bronquial (E). Barras de escala: 50 mm en A; 10 mm en B-E. Tinción: hematoxilina-eosina.
En las vías respiratorias centrales se ha descrito metaplasia escamosa, pérdida de cilios en el epitelio bronquial, hiperplasia de células caliciformes, hipertrofia más hiperplasia y dilatación ductal de las glándulas submucosas, y aumento de la masa de músculo liso83-89. Una medición tradicional del aumento glandular bronquial es el índice de Reid90 o ratio del espesor glandular con respecto al espesor de la pared. Aunque la fibrosis subepitelial o el aumento de espesor de la membrana basal no se ha considerado clásicamente un cambio característico de la EPOC, se ha observado en un subgrupo de pacientes que presentan infiltración inflamatoria eosinofílica22. Aparte de este grupo en particular, se ha descrito la presencia de fibrosis subepitelial en la EPOC91, aunque faltan datos cuantitativos sobre su significación.
Alteraciones estructurales del parénquima pulmonar
El enfisema pulmonar, definido como un agrandamiento de los espacios aéreos distales a los bronquiolos terminales, debido a la destrucción de los tejidos que constituyen sus paredes92, es un fenómeno presente en la EPOC, con un espectro de intensidad y extensión ampliamente variable. Si uno de los extremos de este espectro es el enfisema establecido grave, el extremo opuesto puede estar representado por roturas de las paredes alveolares, que se pueden cuantificar mediante morfometría en pulmones de fumadores sin enfisema identificable, y que son probablemente lesiones precursoras93. Aunque clásicamente se ha incorporado el término ausencia de fibrosis a la definición de enfisema, este concepto ha sido cuestionado por datos que indican que el proceso destructivo del enfisema coexiste con fibrosis de las paredes alveolares circundantes, con un aumento neto de la masa de elastina y colágeno94. Morfológicamente, se han definido 2 formas de enfisema, según el patrón de destrucción tisular95. En una de ellas, la destrucción se focaliza en las zonas proximales de los acinos, en los bronquiolos respiratorios y las zonas más adyacentes del parénquima. Como resultado, en el examen microscópico de las secciones pulmonares, la destrucción se observa localizada en las zonas centrales de los lobulillos, rodeada de zonas de parénquima preservado. En consecuencia, este patrón de enfisema se ha denominado acinar proximal o centrilobulillar. Macroscópicamente, esta forma de enfisema suele afectar de forma más grave a los lóbulos superiores. En la otra forma de enfisema existe una destrucción generalizada de las paredes alveolares y los bronquiolos respiratorios; se denomina panacinar o panlobulillar, y tiene tendencia a afectar de forma preferente a los lóbulos inferiores. Los fumadores pueden presentar ambos patrones de enfisema, pero la forma centrilobulillar es la más característica. El enfisema panlobulillar es, por otra parte, característico de la enfermedad por déficit de alfa-1 antitripsina y comienza a desarrollarse en edades tempranas.
En la EPOC, el enfisema contribuye a la limitación al flujo aéreo, mediante la actuación conjunta de 2 mecanismos: la reducción de la retracción elástica del pulmón que afecta a la capacidad para producir presiones generadoras de flujo, y la reducción de la carga que se opone al cierre de las vías respiratorias, al destruirse los anclajes de las paredes alveolares. Laennec96 fue, probablemente, el primero en sugerir, a principios del siglo xix, que las vías respiratorias periféricas eran el lugar primario de obstrucción en el enfisema, y que la disminución de la retracción elástica del pulmón contribuía a la limitación al flujo aéreo. Las alteraciones mecánicas en una y otra forma de enfisema difieren97. En el enfisema panacinar, la compliance pulmonar es mayor y su incremento se correlaciona con la reducción del flujo aéreo, que parece ocurrir primariamente en función de la pérdida de retracción elástica del parénquima. En el enfisema centrilobulillar, el componente inflamatorio de las vías respiratorias periféricas es mayor y puede ser el principal determinante de la pérdida de flujo aéreo, con la que se correlaciona.
La descripción del entramado de fibras elásticas del pulmón y su destrucción en el enfisema98 supuso un avance significativo en el conocimiento sobre su patogenia hace ya 100 años. Sin embargo, el conocimiento sobre los mecanismos biológicos subyacentes que conducen a la destrucción de tejidos de los acinos pulmonares es hoy un campo en desarrollo. La asociación del déficit homocigótico de alfa-1 antitripsina con enfisema pulmonar26,27 y la creación de modelos animales de inducción de enfisema mediante la instilación de proteasas99,100, generaron la teoría "proteasa-antiproteasa", cuyo alcance explicativo es, sin embargo, limitado, siendo probablemente la digestión por proteasas un mecanismo que contribuye al desarrollo del enfisema en un contexto más complejo. Más de 2 tercios de los fumadores no desarrollan enfisema, y tampoco los pacientes con otras enfermedades inflamatorias pulmonares, en general. Asimismo, la mera digestión de componentes elásticos de la matriz extracelular no es explicación suficiente de la desaparición íntegra de tejidos del parénquima incluyendo su componente celular, el compartimiento microvascular y otros constituyentes intersticiales diversos. La implicación de las células T CD8+ en la EPOC5,19,20,28,29 sugirió un papel de la función citotóxica de estas células en la patogenia del enfisema, una idea que se vio reforzada por el hallazgo de apoptosis de células de las paredes alveolares en fumadores en correlación con los paquetes/año fumados, con un significativo incremento de las células apoptóticas en los sujetos con enfisema, y en paralelo con el aumento de células T CD8+ en el parénquima pulmonar42. Otros habían observado apoptosis de células endoteliales y neumocitos asociada a un aumento de gelatinasas y colagenasas10, y a una disminución de la expresión de factor endotelial de crecimiento vascular (VEGF) y su receptor VEGFR2101, lo que indica, respectivamente, que alteraciones de la matriz extracelular y un defecto de factores de mantenimiento endotelial podrían participar en la inducción de apoptosis en el enfisema. Esta última observación recuperó una antigua hipótesis, según la cual la desaparición de los septos alveolares sería inducida por una previa regresión vascular, ya que las paredes alveolares en el enfisema centrolobulillar son prácticamente avasculares102, y se acompañó de la demostración en un modelo animal de que la señalización a través del receptor de VEGF es requerida para el mantenimiento normal de las estructuras alveolares, y que la inhibición de este receptor induce apoptosis de células septales y desarrollo de enfisema103. Definitivamente, la apoptosis de células estructurales del parénquima pulmonar es con toda probabilidad un mecanismo de importancia central en la "desaparición" de tejido que conduce al desarrollo de enfisema, y la evidencia aportada desde distintos ángulos sugiere que la apoptosis es inducida a través de diversas vías que se pueden complementar. La asociación entre el aumento de células T CD8+ y la apoptosis de neumocitos es particularmente interesante y ha generado la hipótesis del enfisema como enfermedad con un componente autoinmunitario36-38. La investigación presente y futura en esta línea podría aportar importante información sobre los mecanismos de la enfermedad y apuntar hacia nuevas dianas terapéuticas para ralentizar su progresión.
Alteraciones vasculares
En pacientes con EPOC en estadios avanzados, es común la hipertensión pulmonar, que no es explicada simplemente por vasoconstricción hipóxica, ya que persiste al corregirse terapéuticamente la hipoxemia. El hallazgo de inflamación y remodelación en arterias de la circulación pulmonar de sujetos fumadores y con EPOC104 proporcionó un plausible sustrato fisiopatológico de la hipertensión pulmonar en estos pacientes. En arterias pulmonares de pequeño calibre se observó engrosamiento de la íntima y de la capa muscular de la media, y estas alteraciones se correlacionaron significativamente con el grado de enfermedad de pequeñas vías respiratorias y con el grado de enfisema, lo que indica que las alteraciones estructurales en la circulación pulmonar evolucionan en paralelo con los cambios patológicos en las vías respiratorias y el parénquima. Otros estudios posteriores fueron consistentes con estos hallazgos y constataron la existencia de remodelación vascular pulmonar en la EPOC105-109. Algunos de estos estudios mostraron una correlación entre el engrosamiento de la íntima y el grado de infiltración inflamatoria en las vías respiratorias periféricas106,108, lo que indica que la remodelación vascular puede ser inducida por la inflamación. El grado de infiltración inflamatoria directa de las arterias, a base en gran parte de células T CD8+, se correlacionó también con las alteraciones estructurales y con una capacidad disminuida de relajación frente a adenosín difosfato (ADP) de explantes quirúrgicos in vitro109. La disfunción endotelial relacionada con la exposición al humo del tabaco que se ha propuesto en la EPOC como uno de los mecanismos inductores de destrucción de los tejidos parenquimatosos101,103, podría también participar, junto con la inflamación, en la remodelación vascular110.
Sumario y conclusiones
Los factores que determinan la susceptibilidad del desarrollo de EPOC son un frente abierto de investigación. La inflamación, más el estrés oxidativo inducido por la exposición al humo del tabaco y amplificado por la propia inflamación, desempeñan un papel de importancia obvia en la patogenia de la enfermedad. Este papel se desarrolla, a su vez, en combinación con un trasfondo de determinantes genéticos de susceptibilidad, de naturaleza poligénica compleja. La gran variedad fenotípica de la EPOC complica adicionalmente la comprensión de su patogenia. Las células inflamatorias tradicionalmente situadas en el centro de importancia en los mecanismos de la enfermedad son los macrófagos y los neutrófilos. Su papel en la enfermedad es, sin duda, significativo, dado que forman parte de la primera línea de defensa inmunitaria celular frente a agentes dañinos externos, y tienen capacidad de liberar mediadores inflamatorios diversos, radicales oxidantes y una variedad de enzimas proteolíticas con potencial para producir degradación tisular. En la última década, las investigaciones sobre los mecanismos inflamatorios en la EPOC han implicado a la rama adaptativa del sistema inmunitario, y se ha postulado un componente de reacción autoinmunitaria frente a antígenos liberados por daño celular, o generados mediante la modificación química de proteínas por reactantes del humo del tabaco y estrés oxidativo. La evidencia acumulada durante estos años muestra que los agentes efectores de esta reacción autoinmunitaria son células T CD8+ con función citotóxica y capaces de inducir apoptosis de células estructurales de los tejidos pulmonares, y células T CD4+ con perfil de activación tipo Th1, para las que una función plausible es la estimulación de las respuestas citotóxicas a cargo de las células T CD8+. Junto con la respuesta inflamatoria, el estrés oxidativo puede infligir daño directo de diversa índole, y generar bucles de amplificación de la inflamación y de la degradación proteolítica de tejidos. Entre los efectos del estrés oxidativo, un aspecto de especial interés es su capacidad para alterar la regulación de la expresión genética, interfiriendo con los sistemas de remodelación de la cromatina, en concreto con la desacetilación y la acetilación de histonas. Como resultado, se libera la transcripción de genes de acción inflamatoria y se bloquea una vía crítica de acción de la medicación esteroidea. Finalmente, esta diversidad de mecanismos converge en la inducción de cambios estructurales patológicos en los tejidos pulmonares, o remodelación. Estos cambios afectan a las vías respiratorias centrales y periféricas (aumento de la masa de músculo liso, fibrosis, hipertrofia e hiperplasia glandular con hipersecreción de moco), al parénquima (enfisema) y a los vasos pulmonares (enfermedad hipertensiva) y causan en gran parte las manifestaciones clínicas de la enfermedad.







